過渡金屬參與的不對稱催化氫化反應是獲得手性分子最高效的途徑之一,而雜環化合物的不對稱氫化一直以來都是該領域的研究熱點。特別是對于苯并雜環類化合物,其很強的穩定性導致氫化反應條件較為苛刻,而剛性結構更是影響了底物與催化劑的有效配位,其高效不對稱催化氫化更是成為該領域的挑戰性課題。上海交通大學張萬斌教授課題組長期從事過渡金屬催化的不對稱催化氫化反應研究,并取得了一些可喜的進展(近三年代表性工作:Nat. Chem. 2022, 14, 920. Nat. Sci. 2021, e10021. Angew. Chem. Int. Ed. 2021, 60, 23602; 2021, 60, 16989; 2020, 59, 5371; 2019, 58, 15767; 2019, 58, 11505; 2019, 58, 7329. Nat. Commun. 2020, 11, 5935)。2021年,該課題組首次利用BridgePhos–Rh催化體系實現了具有剛性結構內酯型香豆素的高效不對稱催化氫化,以高達98%的收率和99.7%的對映選擇性獲得了手性3-氨基二氫香豆素。通過單晶結構發現在不改變其它因素的前提下,僅通過改變配體碳鏈的長度,可方便地調節BridgePhos–Rh配合物中聯苯骨架的二面角,使配位膦原子上的軸向苯環與聯苯骨架上的苯環具有最強的π-π相互作用,從而提供了最為合適的配位環境,并因此取得了優異的不對稱氫化效果(Angew. Chem. Int. Ed. 2021, 60, 23602-23607)。最近,該課題組利用上述建立的BridgePhos–Rh催化體系,成功地實現了剛性3-氨基色酮衍生物的高效不對稱連續催化氫化,機理研究表明該反應是經歷了一條非常規的動態動力學拆分過程(圖1)。圖1 BridgePhos–Rh催化3-氨基色酮衍生物的不對稱連續催化氫化作者以3-苯甲酰胺色酮(1a)作為模板底物進行反應條件優化,確定最優反應條件為:以[Rh(cod)2]SbF6/(S)-C10-BridgePhos為催化劑,二氯甲烷為溶劑,氫氣壓力40 atm,溫度30 ℃,反應24 h。在最優反應條件下,一系列含有不同取代基的3-氨基色酮底物均以高收率和優異的立體選擇性獲得了氫化產物(圖2)。為了進一步了解反應的過程,作者首先開展了反應動力學實驗(圖3)。發現在反應初期原料1a中的C=C雙鍵被快速氫化,生成中間體2a,同時伴隨著少量3a的生成。當反應進行到2 h時,原料1a只剩下16%,中間體2a產率達到最高值59%。隨后2a被快速消耗,直至12 h反應完全。在反應最初的2 h內,中間體2a的ee值變化很小,隨后逐漸降低,說明在該催化體系下(S)-2a中的碳氧雙鍵還原速率快于(R)-2a。在整個反應過程中,3a的ee值沒有明顯的變化。如果將(S)-2a的ee值,通過反應時間的曲線外推到起始0點時,可以得出反應初始生成(S)-2a的ee值約為76%(圖3a)。同樣,將具有吸電子取代基的底物1h應用于上述的動力學實驗,得到了與底物1a相似的結果。從整個動力學控制實驗來看,化合物1h具有很高的反應活性,反應在90 min能夠完全轉化。同樣通過反應時間的曲線外推到0點時,可以得出反應初始生成(S)-2h的ee值約為84%(圖3b)。分析發現,化合物1a第一次氫化生成的2a起始ee值為76%,即(S)-2a與(R)-2a的相對含量分別為88%和12%,或者說反應起始時(S)-2a的含量為88%。根據反應結束后所有異構體的含量計算,生成3a的3-位為S-構型異構體的含量約為94.3%,即反應結束后該異構體的含量較反應起始(S)-2a的含量有了6.3%的提高(94.3% vs 88%)。這一結果說明反應過程中約有6.3%的(R)-2a轉化為(S)-2a。在對1h用同樣的方法進行分析時發現,大概有5.2%的(R)-2h在第二次氫化過程中轉化為(S)-2h。以上結果均說明在1a或1h的不對稱連續催化氫化過程中,第二次氫化可能存在動態動力學拆分過程(圖3c)。

為了進一步闡明動態動力學拆分過程,作者進行了一系列其它控制實驗。首先,在標準反應條件下,對消旋的中間體2a進行氫化。以上述同樣的方式,對反應結束后的各個異構體含量進行分析,可以得出反應過程中有5%的(R)-2a轉化為(S)-2a。而對消旋的2h進行氫化,發現有22%的(R)-2h轉化為(S)-2h。但是在單一構型的2a和2h氫化過程中,均未觀察到消旋化的現象。很明顯,對第一次氫化反應的中間體2來說,只有當兩個對映異構體同時存在時才能發生消旋化(圖4)。

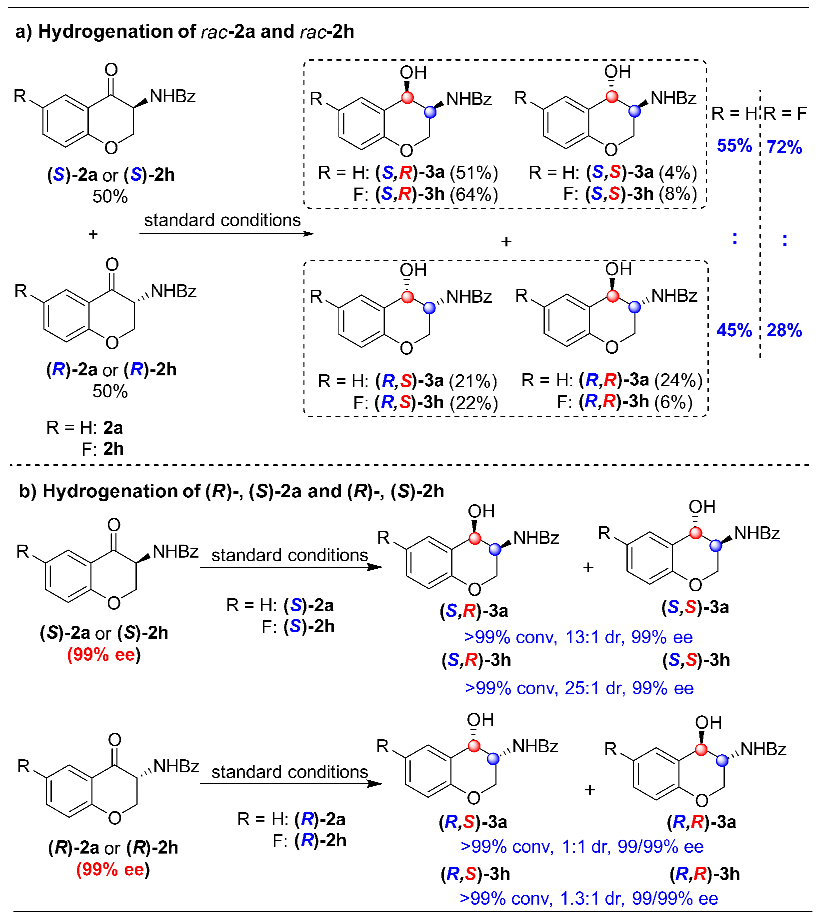
圖4 控制實驗:消旋和對映純1a/1h的不對稱催化氫化為了進一步搞清消旋化過程,作者開展了氘代實驗研究。在標準反應條件下,將摩爾比為85:15的非氘代(S)-2h與氘代(R)-2a混合物應用于上述氫化反應。結果顯示,除了得到氫化產物(S,R)-3h外,也觀察到(S)-2h與氘代(R)-2a有明顯的消旋化現象。需要指出的是,(R)-2a的3-位手性中心上的氘原子沒有任何變化,而由其衍生而來的(S)-2a相應位置上的氘原子卻完全被H原子替代,說明該消旋過程并非是以傳統的烯醇互變形式進行的,很可能是以一種非常規的手性同化方式進行的(圖5)。
圖5 控制實驗:非氘代(S)-2h與氘代(R)-2a混合物的不對稱催化氫化通過在控制實驗的基礎上結合DFT計算,作者提出了反應的可能機理(圖6)。第一步氫化的具體過程為:首先Rh催化劑經氫氣活化,生成Rh(I)-氫氣絡合物,并與底物配位生成中間體0,然后其通過氫氣分子氧化加成和氫原子遷移插入的協同過程生成中間體2,最后中間體2經還原消除得到中間體3。此后,中間體3可以與另一分子氫氣結合并再次發生氧化加成生成中間體4。隨后,4能夠通過配位解離平衡釋放出(S)-2a或者(R)-2a。DFT計算發現,(S)-2a可以繼續發生H遷移插入,生成中間體5a,并接著與氫氣結合,發生氫氣異裂,氫質子轉移到氧原子上生成中間體6a,最后解離出產物,完成第二次氫化過程。然而計算表明(R)-2a在進行第二次氫化時需要很高的活化能,極大地阻礙了其二次氫化反應的進行。因此(R)-2a更傾向于經過一個消旋化途徑轉換為(S)-2a從而完成二次氫化。其具體消旋化過程為:(R)-2a首先與中間體5a結合,生成六配位的銠中間體7a。由于中間體7a的醇陰離子具有較強的堿性,可以攫取(R)-2a立體中心上的氫原子,并使得(R)-2a的手性中心可以通過烯醇型中間體8a發生消旋化。考慮到導向基團的羰基與中心金屬Rh的配位解離平衡,8a和8b可以通過中間體8c相互轉換。最后,中間體8b中羥基的質子發生轉移而形成7b,并解離生成中間體5a和(S)-2a,實現了(R)-2a到(S)-2a的單向轉化。該過程表現為一個手性同化過程,而不是傳統的消旋化過程。

接下來作者進行了該不對稱連續氫化反應的放大實驗,在底物/催化劑最高為500/1的摩爾比下,反應仍可高收率、高立體選擇性地合成手性3-氨基-4-色烷醇,該產物可以被進一步衍生轉化為多種藥物活性中間體(圖7)。

該氫化反應的中間體(S)-2a(可由3a氧化而得到)也是手性新藥物開發過程中非常有用的合成砌塊,通過一系列轉化可得到多樣化的手性天然產物或臨床新藥(圖8)。

以上成果近期發表在國際著名期刊《美國化學會雜志》(Journal of the American Chemical Society, 2022, DOI:10.1021/jacs.2c09266)上,上海交通大學張萬斌教授為該論文的通訊作者,上海交通大學藥學院博士生徐運楠和上海交通大學化工學院博士生羅亦聰為共同一作,該項工作主要得到了國家自然科學基金,國家重點研發項目基金和上海市教委基金的資助。


 在線客服
在線客服
 快速發布
快速發布
 我的店鋪
我的店鋪
 我的化學加
我的化學加
 我的消息
我的消息
 我要充值
我要充值


 回到頂部
回到頂部















