

 在線客服
在線客服
 快速發布
快速發布
 我的店鋪
我的店鋪
 我的化學加
我的化學加
 我的消息
我的消息
 我要充值
我要充值


 回到頂部
回到頂部



買產品

克難題

技術供需

求職招聘

發定制

項目整合

園區推薦

行業資訊

化學加智庫

商家
買產品
克難題
技術供需
求職招聘
發定制
項目整合
園區推薦
行業資訊
化學加智庫
商家
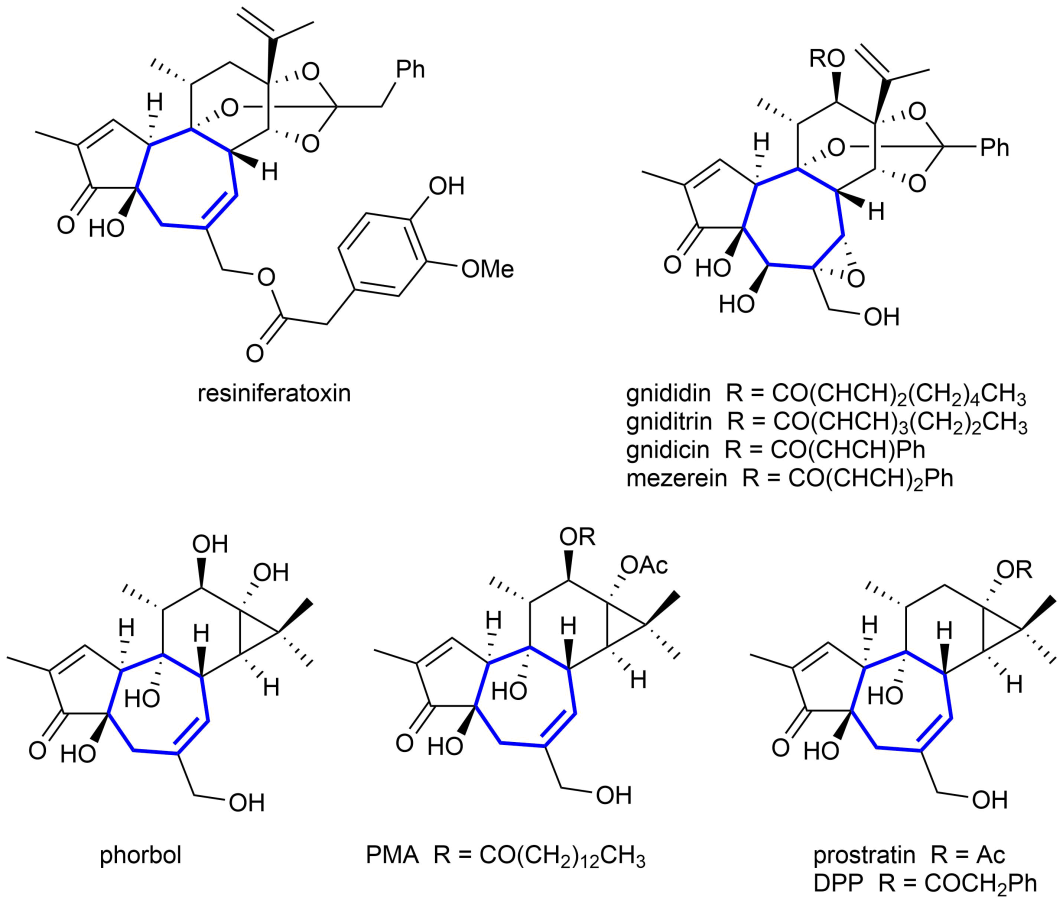
圖1. 一些含有七元碳環結構的天然產物。
1,2-二乙烯基環丙烷的Cope重排是一種合成七元碳環的方法,但是底物的合成通常較為繁瑣。為了解決這一問題,合成化學家后來發展出了過渡金屬催化的串聯環丙烷化/Cope重排反應,實現了6-7和5-7雙環體系的高效構筑(圖2a)。受到這些工作的啟發,北京大學余志祥課題組于2014年發展了一種金催化的雙烯雙炔環化異構化反應(該反應亦可被稱為形式[4+3]環加成/碳氫鍵活化反應),通過巧妙地引入1-乙烯基-2-炔基環丙烷的Cope重排反應,高非對映選擇性地合成了具有七元碳環的6-7-5三環化合物(圖2b)。作者利用金催化的串聯環丙烷化/Cope重排/碳氫鍵活化反應,依次構建了三根碳碳鍵和三個環,從而實現了由簡單線性分子到復雜多環分子的高效轉化。此外,通常情況下惰性烷基碳氫鍵活化需要相對苛刻的反應條件,而在此工作中碳氫鍵活化反應在室溫下即可順利發生。
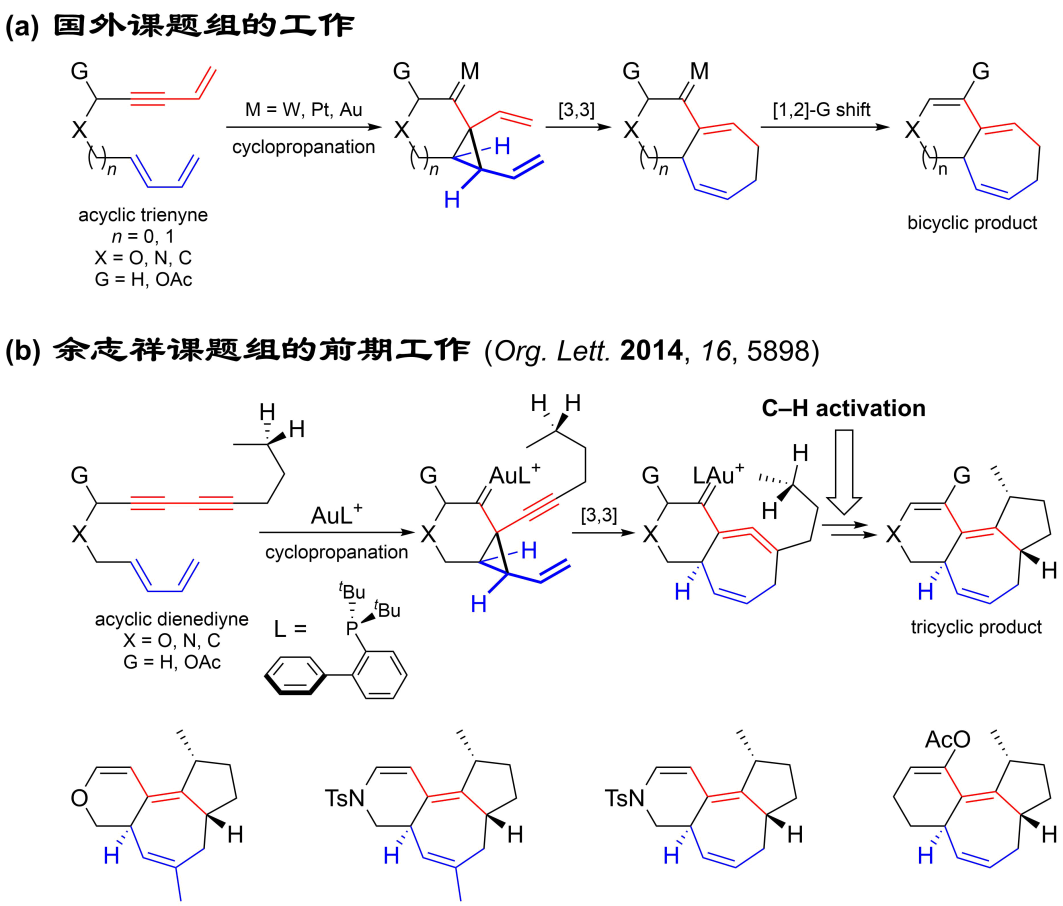
圖2. (a)串聯環丙烷化/Cope重排反應;(b)串聯環丙烷化/Cope重排/碳氫鍵活化反應。
最近,余志祥課題組又對該反應的機理進行了研究。密度泛函理論計算和氘代實驗表明該反應經由配體交換、環丙烷化、1-乙烯基-2-炔基環丙烷的Cope重排、碳氫插入和[1,2]-氫遷移等基元步驟完成(圖3)。其中,反應的決速步是環丙烷化步驟。通常情況下,通過1-乙烯基-2-炔基環丙烷的Cope重排得到的七元環聯烯在合成上的價值不大,這是由于其可以發生后續的二聚反應。而在余志祥課題組的工作中,七元環聯烯C具有一個烯基正離子共振式D。烷基碳氫鍵的活化正是通過該烯基正離子對碳氫鍵進行快速插入予以實現的(理論計算結果表明這一步的活化Gibbs能僅為1.5 kcal/mol)。
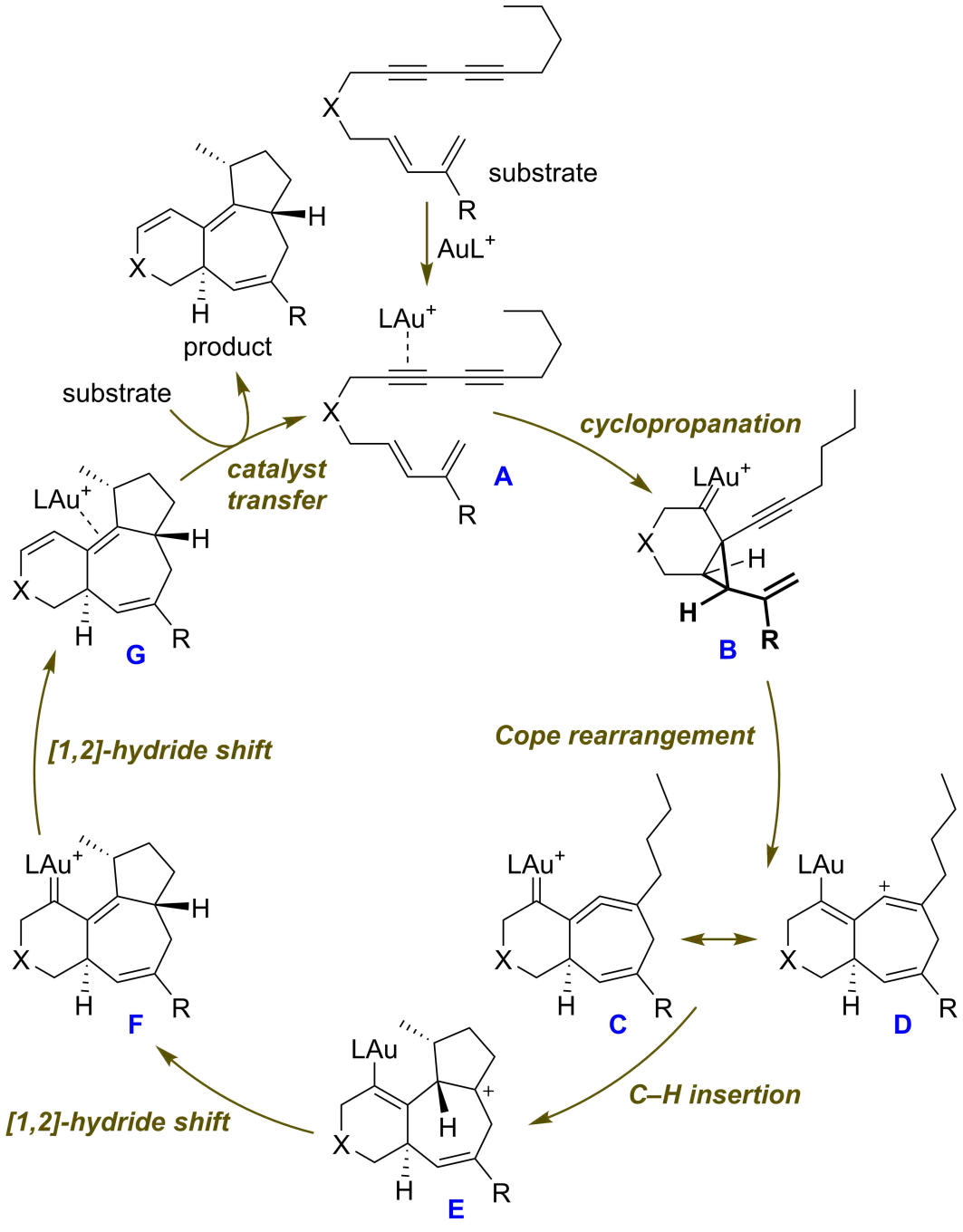
圖3. 催化循環。
上述反應和機理適用于X為O或NTs的底物(當X為CH2且遷移基團G為OAc時,機理有相應的變化),而不適用于雙酯橋底物(當X為C(CO2Me)2且遷移基團G為H時)。這是由于氧橋和氮橋底物的環丙烷化具有endo-dig選擇性(圖4a,左),從而能夠引發后續的Cope重排;而雙酯橋底物的環丙烷化具有exo-dig選擇性(圖4a,右),不能引發后續的Cope重排。基于此,作者提出了一個設想:如果使用縮碳底物,則exo-dig環化將生成具有高度張力的4-3并環骨架,從而得到抑制。作者進一步的密度泛函理論計算支持了這一觀點:相對endo-dig環化而言,exo-dig環化在動力學上較為不利。基于這些理論預測,作者隨后在實驗上成功地實現了縮碳底物的環化異構化反應,為構建具有全碳骨架結構的5-7-5三環和5-7-6-6四環化合物提供了新的合成工具(圖4b)。在這個新的環化異構化反應中,2a的合成仍然通過烷基碳氫鍵活化實現,而在2b-d的形成過程中則改用了Friedel-Crafts反應。這一新反應的實現很好地展示了余志祥課題組理論與實驗相結合的研究理念在解決合成化學實際問題中的優勢。
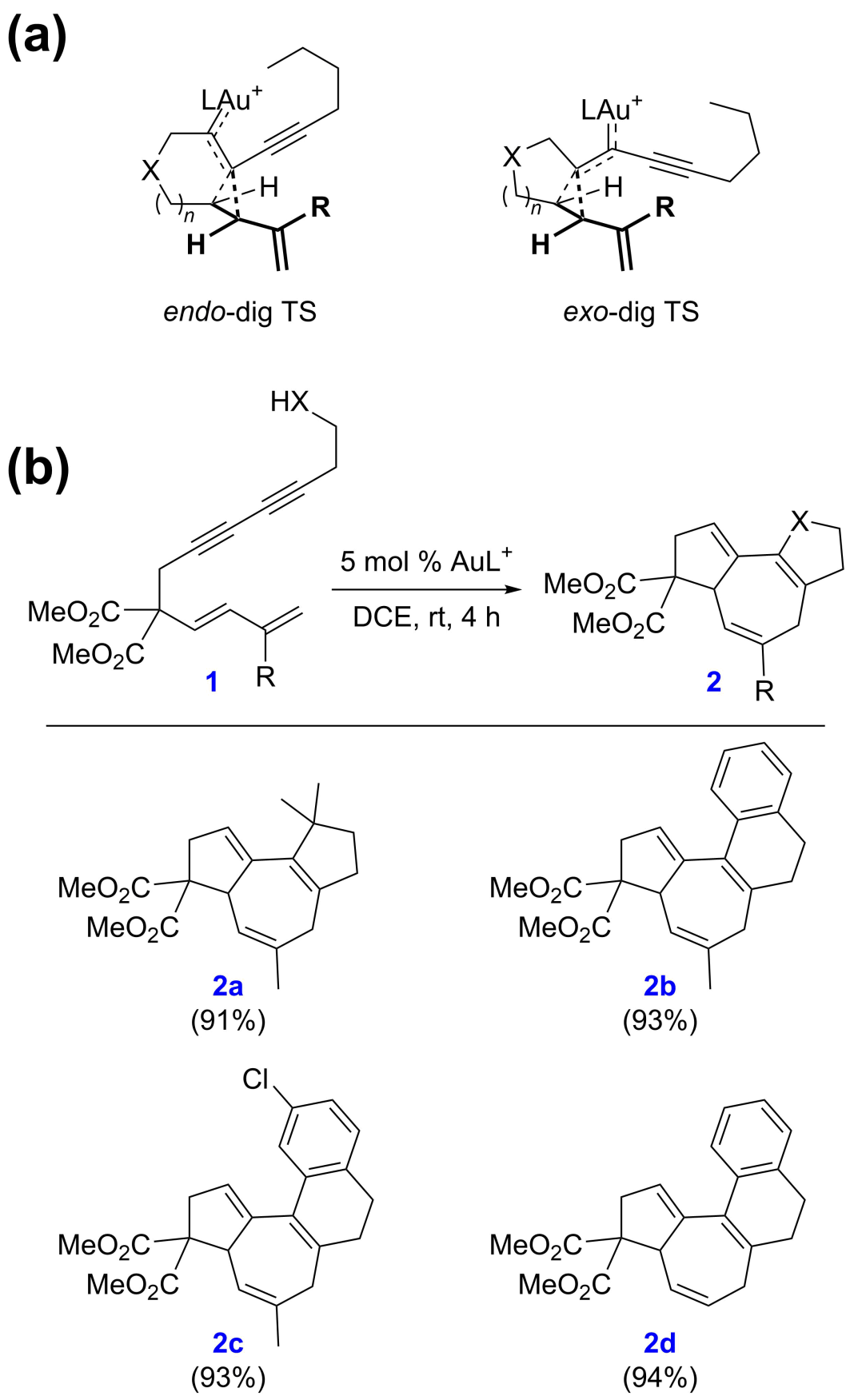
圖4. 通過機理研究設計新的環化異構化反應。
該課題的前期工作(合成方法學部分)由余志祥課題組的蔡沛君博士、王熠博士和柳成航博士完成,發表在Org. Lett. 2014, 16, 5898上。最近的機理研究工作由王熠博士和蔡沛君博士完成,發表在J. Am. Chem. Soc. 2020, 142, 2777上(https://pubs.acs.org/doi/10.1021/jacs.9b10362)。 該工作得到國家自然科學基金委重點基金“金屬催化環加成反應機理研究和反應發展”(基金號,21933003)的支持。
聲明:化學加刊發或者轉載此文只是出于傳遞、分享更多信息之目的,并不意味認同其觀點或證實其描述。若有來源標注錯誤或侵犯了您的合法權益,請作者持權屬證明與本網聯系,我們將及時更正、刪除,謝謝。 電話:18676881059,郵箱:gongjian@huaxuejia.cn
