

 在線客服
在線客服
 快速發布
快速發布
 我的店鋪
我的店鋪
 我的化學加
我的化學加
 我的消息
我的消息
 我要充值
我要充值


 回到頂部
回到頂部



買產品

克難題

技術供需

求職招聘

發定制

項目整合

園區推薦

行業資訊

化學加智庫

商家
買產品
克難題
技術供需
求職招聘
發定制
項目整合
園區推薦
行業資訊
化學加智庫
商家
自1994年以來,化學家們從苔類植物中分離和表征了一組具有并環呋喃酮的非常復雜的二萜化合物,如pallavicinin (1)、pallavicinin (2)和pallambins A-D(3-6)(圖1A),它們含有4-6個擁擠環和7-10個連續手性中心,包括1-2個全碳季碳中心,其全合成極具挑戰。然而,這類天然物并沒有顯示出顯著的生物活性,可能是因為它們自然來源極少,從而限制了其生物評價。因此,對這些二萜類化合物進行簡明的化學合成可以提高這些分子的可用性,以便進行全面的生物評估。
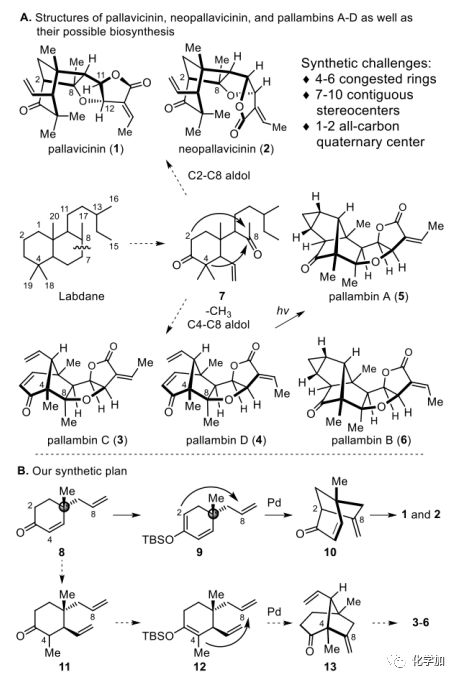
圖1. 目標分子的結構、生源合成及合成計劃(圖片來源:Angew.Chem. Int. Ed.)
Asakawa提出了這些天然產物的合理生物合成途徑(圖1A)。labdane型二萜類化合物C7-C8鍵的斷裂提供了共同的前體7。1和2是通過分子內Aldol反應重建C2-C8鍵而產生的。3和4是通過去甲基化和C4-C8鍵重建形成的。婁紅祥等人進一步證明了3和4通過光誘導的雙自由基重排到5和6的相互轉化。
1-6全新且極具挑戰性的分子結構吸引了合成化學家的廣泛關注,黃乃正課題組率先從Wieland-Miescher酮合成了(±)-1至(±)-4。Carreira等人在2015年以Diels-Alder反應實現了 (±)-5和(±)-6的首例全合成。同年,賈彥興課題組從已知的手性環己烯酮8(圖1B)出發完成了(-)-1和(+)-2的不對稱合成。2016年,Baran課題組報道了一種優雅的(±)-3和(±)-4的合成,且沒有使用保護基。
由于作者最近成功地從8合成了(-)-1和(+)-2,并受上述生物合成假說啟發,作者設想8可以作為合成1-6的公共中間體。化合物8很容易轉化為12,經Pd-催化氧化環化得到[3.2.1]雙環酮13,13可轉化為3-6(圖1B)。因此,可以實現1-6的多樣性合成。在此,作者報道了在不使用保護基的情況下,第一次不對稱合成pallambins A-D(3-6)。
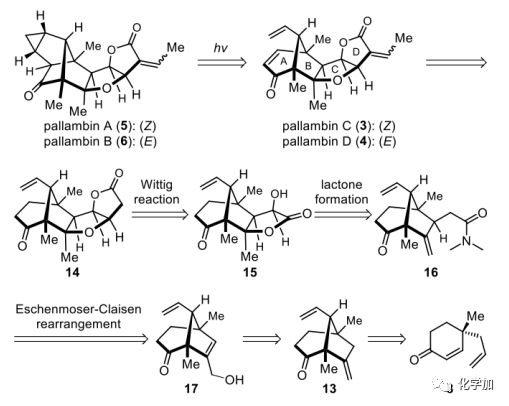
圖2. pallambins A-D的逆合成分析(圖片來源:Angew.Chem. Int. Ed.)
3-6的逆合成分析如圖2所示。作者設想5和6可以通過婁紅祥的3和4的光誘導相互轉化很容易地得到。反過來,3和4可以從四環化合物14得到,其中BCD三種不同的環系逆推至三種不同的環化方式。因此,D環由內酯15的分子內Wittig反應而形成。C環是由酰胺16形成內酯形成的,酰胺16可以通過烯丙醇17的Claisen重排而生成,烯丙醇17可以從雙環化合物13很容易地合成,13可以從8得到。
全合成從已知的手性化合物8開始,8可以按Stoltz的方法以84% ee輕易制備。乙烯基與8共軛加成,然后甲基化,得到酮11,在C5處的非對映體比(dr)為3:1。用Et3N和TBSCl對酮11進行處理,得到相應的熱力學TBS烯醇醚,再在優化的Pd-催化氧化條件下,得到了期望的雙環[3.2.1]辛烷體系13,產率64%。用m-CPBA對13的富電子雙鍵進行化學選擇性環氧化,得到相應的環氧化產物,在1,3-二甲基咪唑啉-2-酮(18)存在下用PTSA處理,得到所需的烯丙醇17。穩健得到17以后,接著集中于C環的構建。在甲苯中115 °C下加熱17和N,N-二甲基乙酰胺二甲基縮醛,以單一對映體的形式得到了理想的γ,δ-不飽和酰胺16,產率為88%。在回流乙醇中用H2SO4處理16,得到內酯19,收率84%。
為了在C11處安裝羥基,必須保護19中的酮。然而,對分子模型的進一步觀察表明,由于空間擁擠的A/B環系屏蔽了C3-酮基的α-位,游離C3-酮基可能不會影響α-羥基化反應。因此,作者大膽地選擇直接引入C11羥基,而不保護C3酮。在對幾種條件進行測試后,作者發現,用3當量LiHMDS對19去質子化,再加入戴維斯試劑(20),得到a-羥基內酯21,為單一的立體異構體。由于19的高張力籠狀結構,羥基化反應只發生在位阻較小的凸面上,與C8甲基成順式。這與天然產物所需的立體化學是相反的。因此,采用氧化/還原策略對C11羥基的構型進行了翻轉。用DMP氧化21得到相應的酮內酯,并用LiAlH(Ot-Bu)3進行了完全的化學和立體選擇性還原,得到所需的醇15為單一立體異構體,收率為99%。
得到了三環化合物15,接著引入最后一個環系。在間二甲苯中160 ℃下用(三苯基磷酰亞甲基)烯酮和15反應,完成了a-羥基的酰化反應和隨后的分子內Wittig反應,得到了四環酮酯22。然后進行了四環酮酯雙鍵的化學和立體選擇性還原。經過廣泛的實驗,作者發現CuI與Red-Al反應原位生成CuH還原22,得到目標產物14,產率為94%,其結構經X-射線晶體分析確證。
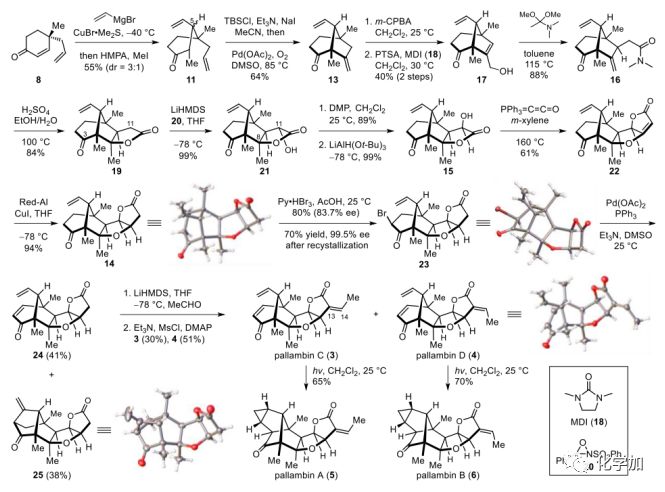
圖3. pallambins A-D的全合成(圖片來源:Angew.Chem. Int. Ed.)
在這個階段,作者需要安裝C1,C2雙鍵;然而,這是非常具有挑戰性的。首先研究了酮直接脫氫的一步法,但是,用IBX氧化酮14,以及Pd-催化脫氫等其他方法,都沒有得到24。最后,14與Py·HBr3在HOAc中溴化得到單一立體異構體23,產率為80%。用Li2CO3/LiBr、DBU或其它堿對23的脫溴化氫反應進行了篩選。反應在低溫(<80 ℃)下不發生,在高溫(>80 ℃)下原料分解,未觀察到預期產物24。這些結果表明,23和(或)24可能對高溫條件下的堿敏感。因此,需要開發一種溫和的脫溴化氫方法。
在Pd-催化Heck反應溫和的反應條件和反應機理的啟發下,作者認為a-溴酮23對Pd(0)物種的氧化加成可以得到相應的烷基鈀物種,經過β-氫消除能得到所需的烯酮24。經過廣泛的試驗,作者發現在優化的條件下(DMSO中的Pd(OAc)2、PPh3和Et3N)處理23,可獲得所需產物24,收率為41%,Heck型產物25的收率為38%。在24上裝上亞乙基,以30%的產率得到目標分子pallambin C(3),51%的產率得到pallambin D(4),合成產物3和4的物理數據與文獻報道一致。
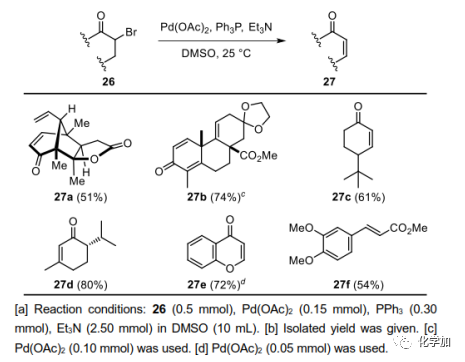
圖4. a-溴代酮脫溴化氫(圖片來源:Angew.Chem. Int. Ed.)
最后,根據婁紅祥課題組的研究,在紫外光下將pallambin C(3)和D(4)轉化為pallambin A(5)和B(6)。有趣的是,在較溫和的條件下(紫外燈,8 W),在25 ℃的CH2Cl2中,3和4的單獨輻照分別產生5和6,而在5和6之間以及3和4之間沒有雙鍵順式/反式異構化。這一結果清楚地表明,在?13(14)處,雙自由基重排所需的能量比順式/反式異構化所需的能量要低。合成產物5和6的物理數據與文獻報道一致。
總結:北京大學賈彥興課題組在不使用保護基的情況下,從已知的環己烯酮8出發,用15-16步反應完成了pallambins A-D的首次不對稱全合成。這種無保護基合成的成功主要取決于幾個高度的化學和立體選擇性反應:Pd-催化氧化環化構建[3.2.1]-雙環結構,Claisen重排/內酯形成序列構建C環,分子內Wittig反應形成D環。在此過程中,作者還開發了一種溫和的a-溴代酮脫溴化氫方法。
撰稿人:詩路化語
聲明:化學加刊發或者轉載此文只是出于傳遞、分享更多信息之目的,并不意味認同其觀點或證實其描述。若有來源標注錯誤或侵犯了您的合法權益,請作者持權屬證明與本網聯系,我們將及時更正、刪除,謝謝。 電話:18676881059,郵箱:gongjian@huaxuejia.cn
