

 在線客服
在線客服
 快速發布
快速發布
 我的店鋪
我的店鋪
 我的化學加
我的化學加
 我的消息
我的消息
 我要充值
我要充值


 回到頂部
回到頂部



買產品

克難題

技術供需

求職招聘

發定制

項目整合

園區推薦

行業資訊

化學加智庫

商家
買產品
克難題
技術供需
求職招聘
發定制
項目整合
園區推薦
行業資訊
化學加智庫
商家
“重磅炸彈”新藥合成與工藝綠色化
Blockbuster Drugs Synthesis and Greening Pharmaceutical Process
張 霽1,2,張英俊1,2,聶 飚1
(1. 廣東東陽光集團藥業研究院,廣東東莞 523871;2. 抗感染新藥研發國家重點實驗室,廣東東陽光藥業,廣東東莞 523871)
摘要:通過評述CDK4/6 抑制劑帕博昔布、丙肝NS3/4A 蛋白酶抑制劑司美匹韋和NS5A 抑制劑Elbasvir 等“重磅炸彈”新藥合成及工藝過程,對其中工藝研究的重點難點和瓶頸反應進行全面分析,揭示關鍵反應的優化以及分離純化節點的選擇實施,是整個工藝綠色化的重點。同時對手性相轉移催化技術、酶催化方法和SFC 分離技術等綠色手段在合成工藝中的應用進行了案例解析。總結出一些基本的工藝特點和規律, 最后展望了工藝綠色化的一些新前沿、新亮點。
關鍵詞:“重磅炸彈”藥物;綠色工藝;工藝優化;藥物合成

1 前言
“重磅炸彈”新藥一般是指年銷售額達10 億美元以上的藥物[ 1],如輝瑞的MG-CoA 還原酶抑制劑立普妥(Lipitor)、艾伯維的阿達木單抗(Humira)以及默沙東的二肽基肽酶-4(DPP-4) 抑制劑西格列汀( sitagliptin) 等。由于這類藥物的作用機制新穎、療效優異,上市后迅速被醫生和患者接受使用,因此獲得的社會及經濟效益都非常可觀。與此同時,這些重磅藥物均由世界一流的制藥企業研發和生產,其開發的合成工藝方法也是非常值得學習和借鑒的。例如西格列汀的化學工藝就曾2 次獲得美國綠色化學的最高獎(Presidential Green ChemistryChallenge Award)[2—4]。本文就新近出現的“重磅炸彈”藥物的合成路線及工藝進行分析,探討新藥合成工藝開發的過程和綠色化的途徑[5]。
2 帕博昔布(palbociclib,17) 的合成與工藝綠色化
17 是全球首個上市的CDK4/6 抑制劑[6]。基于Ⅱ期臨床無進展生存期(progression free survival,PFS) 的優秀療效數據,2015 年被FDA 加速批準,
用于治療HR+/HER2- 晚期乳腺癌,第一年即實現了7.23 億美元的銷售額,2016 年上半年全球銷售額達到9.42 億美元,預計全年銷售額可達23 億美元,已成為名副其實的“重磅炸彈”新藥。該藥物發現的初始路線( 圖1) 是從4- 氯-2- 甲硫基嘧啶-5- 甲酸乙酯(1) 出發,經過7 步反應構建溴代吡啶并嘧啶酮母核中間體10,然后經過特別的氧化劑11 處理,得到了關鍵中間體亞砜12,隨后的親核取代反應連接氨基吡啶13,接著在Pd(PPh3) 4催化下,利用Still 偶聯反應制備了烯醇醚16,經鹽酸酸性水解除去保護基,獲得了17 的鹽酸鹽。該路線共涉及11 步反應,總收率僅4.8% [7]。
經過考查和前瞻性分析,不難發現該路線有以下不足和缺陷:①該路線需使用易燃易爆的氫化鋁鋰(LiAlH4),以及純度和品質無法確定、轉移困難和有加料風險的氫化鈉(NaH);②中間體多次不合理的氧化態改變(LiAlH4 還原-MnO2 氧化- 格氏試劑加成-NMO/TPAP 氧化- 硫醚的特殊氧化成亞砜等);③采用非綠色的Horner-Wadsworth-Emmons反應用來構建吡啶并嘧啶酮雜環母核;④ Still 偶聯反應使用有毒的錫試劑,殘留錫不易徹底消除,而且會產生極性和溶解度與產物相似的雜質,難以分離純化;⑤需要柱色譜分離純化中間體7、12 和16等,難以實現大規模工業化生產;⑥值得一提的是,罕見的氧化劑即希夫堿環氧化物11 制備極其困難,穩定性極差、易分解,純化工藝及純度都不好控制,儲存也是難題,更麻煩的是,用該試劑制備的亞砜反應活性不高,隨后的親核取代反應產率很低(38% ),嚴格來說該反應是整個過程的第一個瓶頸反應(bottleneck reaction) [8]。
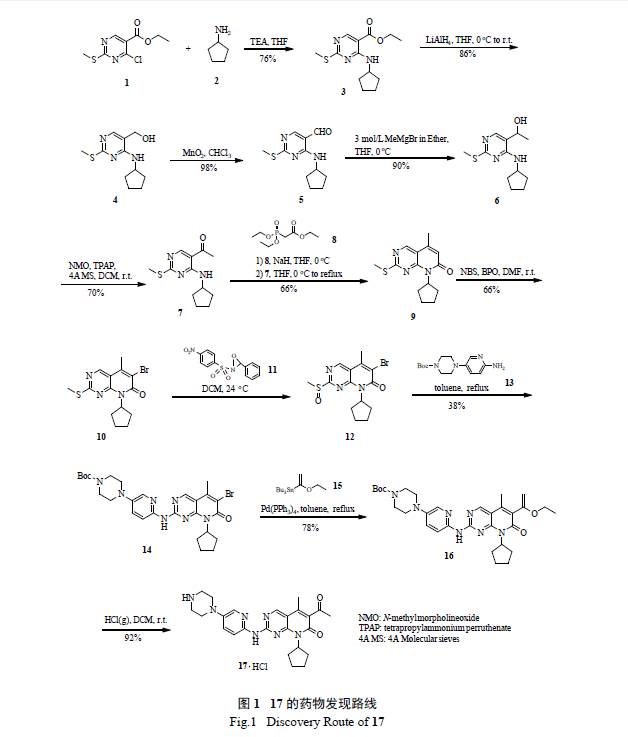
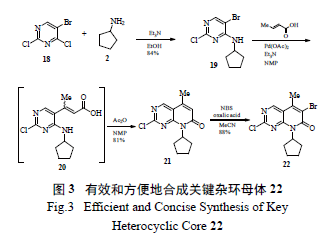
輝瑞的工藝研發者對這個瓶頸反應進行了全面攻關,發現當X=MeSO2 時,親核取代反應有所改善( 產率增至50% ),但優化反應條件后收率仍不理想。考慮到制備化合物10a 和10b 步驟多(10 ~11 步),故決定迅速開發以X=Cl 為中間體的關鍵反應( 圖2),因為該中間體22 僅需4 步就能方便有效地從5- 溴-2,4- 二氯嘧啶(18) 制備( 圖3):①溫和條件下的選擇性胺化;②區域選擇性Heck 反應;③ NBS 溴代反應得到溴代吡啶并嘧啶酮母核中間體[9]。
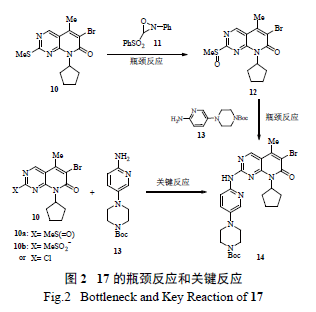
隨著化合物22 的成功制備,氨基吡啶和化合物22 的高度區域選擇性SNAr 偶聯便成了關鍵反應(key reaction)。輝瑞工藝研究人員通過系統的研究發現,許多文獻報道的鈀催化C-N 偶聯反應在此處的應用并不理想,通過篩選不同的鈀鹽[ 例如Pd(OAc)2、Pd2(dba)3、POPd]、膦配體(BINAP、DavePhos、JohnPhos)、堿(NaOt-Bu、K3PO4、Cs2CO3)以及溶劑( 甲苯、DME), 最好的收率也僅為40%。考慮到鈀催化劑成本較高,且實驗結果難以令人滿意,故研究人員認為該法不切實際,難以用于放大生產。
相對于亞砜10a 和砜10b,氯代的吡啶并嘧啶-7- 酮22 的反應活性很低,且氨基吡啶是一個弱的雙向的親核試劑(ambident nucleophile),在中性條件下即便是在回流的甲苯里,也得不到預計的產物14。相反,在弱堿條件下,卻能得到少量的區域異構體23( 圖4),這也說眀氨基吡啶雜環上的氮親核活性更高。顯然如何將區域異構體23 轉變為理想產物14 也是必須面對和解決的一個難題。

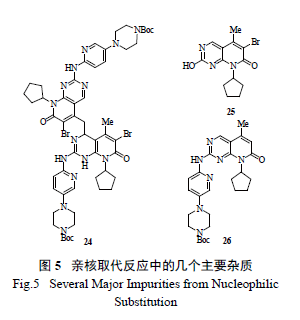
隨后的試驗表明,區域異構體23 在強堿性條件下,通過調節pH 值或淬滅反應,可轉化為理想產物14。欣喜的是,在線紅外(ReactIR) 光譜跟蹤檢測發現,在強堿LiHMDS 作用下,反應物13 能脫質子形成氨基吡啶負離子,能有效制備14,當加入過量的堿(2.5 倍摩爾量) 和13(2.2 倍摩爾量) 時,反應轉化率能從28%提高至85%。美中不足的是,在過量的強堿條件下,14 不穩定,會產生二聚體副產物24( 1,6- 加成產物),給分離純化帶來極大的困擾( 圖5)。如降低堿的用量( 小于2 倍摩爾量),則轉化率低于50%。顯然,從原子經濟學和產物的轉化率、分離純化等多方面考慮,使用LiHMDS 作堿仍不是最佳選擇。
初始的反應動力學研究認為該親核取代反應是一個二級反應( 圖6),這能夠解釋為什么中間體形成后,需要另一分子的鋰化氨基吡啶負離子作進攻試劑。但是隨后的反應轉化率數據和動力學模型DynoChem 計算發現,二級反應的機理和數據不能與實驗結果很好匹配,相反,反應數據卻能和一級反應很好吻合,故認為該反應是經過四元環的過渡態,即經過[1,3] 重排遷移完成的( 圖7)。
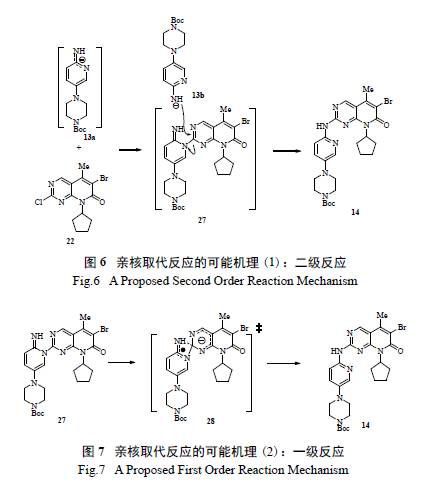
HOMO 的計算也表明,在中性條件下,化合物13a 中吡啶環上的氮和環上的氨基有大致相同的親核活性,許多情況下,產生區域異構體27 顯然是動力學上有利生成的。由于環外的氮有較高的電子云密度,且不易離域化,故通過使用其他的強堿試劑,極有可能形成脫質子的氨基負離子。初步篩選強堿,如NaOtBu, KOtBu, NaOt-Am 可得到約30%~ 40%的理想產物14,但不可避免地帶來水解副產物25。若嚴格干燥各種試劑并小心處理反應,可以有效控制雜質生成,收率能提高到87%,但在大規模的放大生產中,完全嚴格地除水是不現實的。由此,輝瑞研發團隊決定使用氯化異丙基鎂作為堿參與親核取代反應,結果反應進行得非常干凈徹底,且未生成二聚體副產物24。當13 和格氏試劑摩爾比1.1 ∶ 2 時,收率81%;優化后摩爾比調整為1.3 ∶ 2.2 時,收率提高到94%,但有少量脫溴副產物形成。他們通過在線紅外和離線分析發現反應機理存在2 種可能的途徑:(1) 直接親核取代;( 2) 反應機理和前述相同,但可能中間體23 轉化為產物14 太快,無法用在線紅外跟蹤檢測。鑒于用氯化異丙基鎂作堿在反應過程中會產生異丙烷氣圖6 親核取代反應的可能機理(1):二級反應Fig.6 A Proposed Second Order Reaction Mechanism圖7 親核取代反應的可能機理(2):一級反應Fig.7 A Proposed First Order Reaction Mechanism體,在最后的優化工藝中,改用氯化環己基鎂,解決了產生氣體所帶來的安全隱患,已成功應用于實際生產。
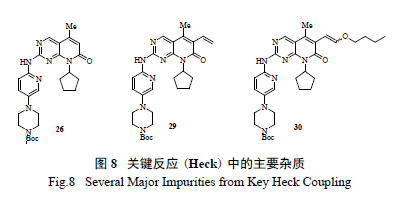
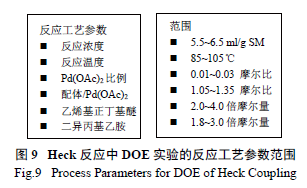
最后,利用鈀催化的Heck 反應引入烯醇醚成為整個工藝的另一個關鍵反應。研發人員通過DOE實驗[10],系統全面地優化了實驗參數,合理化了可操作的實驗空間,進一步抑制和控制了雜質( 圖8) 含量,這包括催化劑的選擇、50 個不同的配體和5 個不同堿的篩選組合,一共進行了250 個反應( 圖9),分別找出了脫溴雜質26 產生的敏感因素[Pd(OAc) 2 與溫度關系圖];乙烯基雜質27 和區域異構體28 產生的敏感因素[DPEphos 配體與Pd(OAc)2 的關系圖],真正將反應的控制從定性的范圍提高至一個定量的高度。優選后的反應將26、乙烯支鏈雜質29 和區域異構體30 控制在合理范圍,同時使這個關鍵反應能夠在一個極為合理的參數空間三維進行操作,而不是一個局部二維的優化。
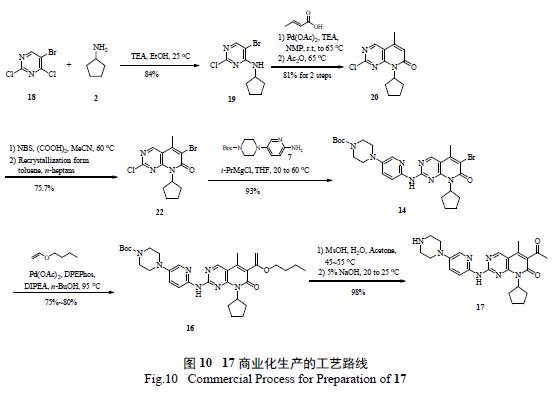
綜上所述,不難發現輝瑞工藝研發人員通過對17 藥物發現路線的認真評估和研究,通過引入更加合理易得的原料,首先迅速克服了瓶頸反應的難題,隨后將2 個關鍵反應( 親核取代和Heck 偶聯) 進行合理分析、優化和動力學研究,找到了合理操作及控制的空間,避免了雜質的大量形成,簡化了反應的后續分離純化,解決了一系列安全隱患。將反應從原來的11 步簡化到可商業化生產的7步( 圖10),總收率也從初始的4.8%改善到目前的37.7% [11]。總而言之,整個工藝的綠色化表現在:①合成策略和關鍵中間體的正確選擇;②非綠色、不安全反應的棄用;③現代PAT 技術和動力學研究輔助工藝優化;④ DOE 實驗優化反應;⑤ Heck 反應置換Still 偶聯反應;⑥避免不合理的氧化還原反應過程。
3 丙肝NS3/4A 蛋白酶抑制劑司美匹韋的合成及工藝綠色化
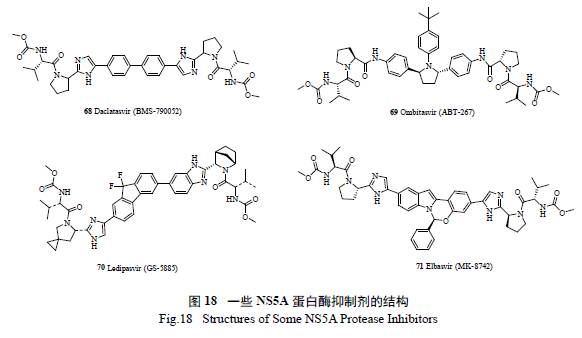
2014 年上市的吉利德史詩級“ 重磅炸彈”NS5B 抑制劑索非布韋( sofosbuvir) 以優異的治療效果、廣譜的抗病毒活性、高耐藥屏障以及極低的交叉耐藥可能性,迅速占領了市場,2015 年全年銷售53 億美元,sofosbuvir 和ledipasvir 的復方Harvoni 全年達139 億美元,創造了銷售奇跡[ 12]。楊森制藥開發的司美匹韋( simeprevir) 是第二代NS3/4A 抑制劑,它的出現對NS3/4A 抑制劑的開發是革命性的,不僅表現出對NS3/4 蛋白酶極高的抑制活性[13],而且臨床上與干擾素/ 利巴韋林合用的三聯療法治愈率極高,12 周的持續病毒響應時間高達88.6%,相對之前的療法有了極大提高,同時治療周期也縮短到12 周。2014 年也創下了年銷售23億美元的佳績。其他一些丙肝NS3/4A 抑制劑的結構見圖11。
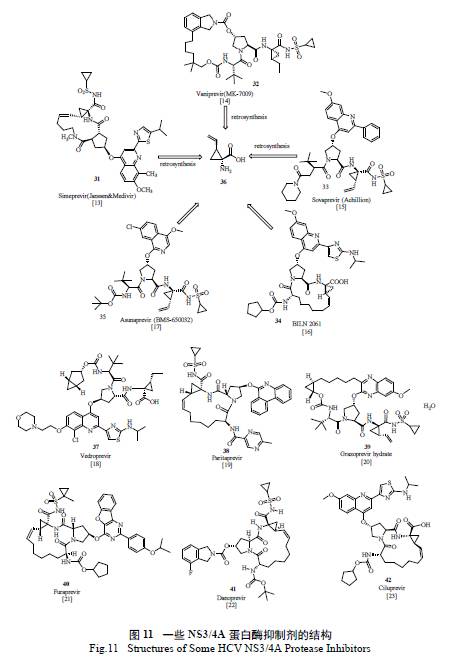
從圖11 來看,這些抑制劑都含有一個共同的手性小分子,即保護的(1R,2S) -1- 氨基-2- 乙烯基環丙基羧酸,Boehringer-Ingelheim(BI) 的化學家首先發展了一種以價廉易得的甘氨酸酯鹽酸鹽為起始原料的簡約方法( 圖12):選擇1,4- 二溴代-2- 丁烯為另一原料,利用串聯的SN2-SN2' 雙烷基化過程,一步到位極為便利地制備了外消旋的氨基酸。值得一提的是,作為關鍵反應,反應條件的選擇和優化對控制主要副產物48 的形成至關重要。通過數十個反應條件的仔細篩選,將理想產物45 與48 的比例從極不理想的1.2 ∶ 1 優化至>40 ∶ 1,易于分離純化,也為疊縮工藝(Telescoping Process) 的實施打下了堅實的基礎[24]。
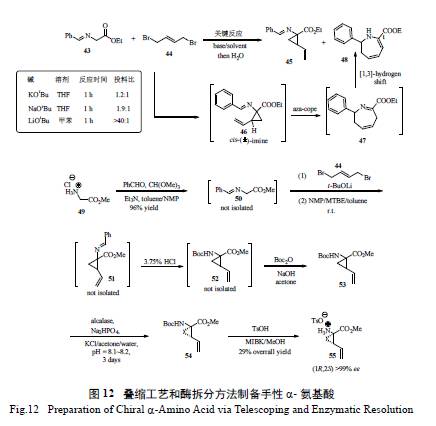
從工藝綠色化的角度來分析整個流程,不難發現該工藝對每個基元反應的研究和優化都很透徹,雖然前面數個反應的產物、副產物和原料都是液體,但通過反應的高產率、高效輔以理想的中控,避免了3 個液體中間體的分離純化,實現了疊縮工藝的完美結合,成功完成了從公斤級實驗到中試的跨越。而利用酶催化選擇性在溫和條件下拆分外消旋氨基酸,也是綠色工藝的很好體現。
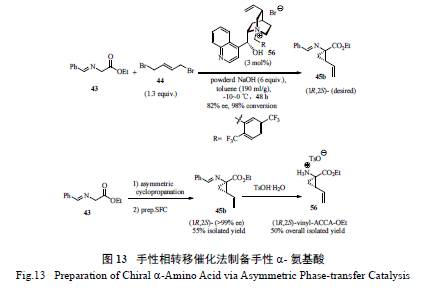
在BI 的工作基礎上,Merck 的工藝化學家巧妙使用了無需過渡金屬催化的手性相轉移催化綠色工藝,用以甘氨酸希夫堿和二溴丁烯為原料的合成方法[25],一步到位構筑了含有環丙烷基和乙烯基的手性α- 氨基酸酯( 圖13)。值得一提的是,通過對反應條件的篩選和優化( 手性相轉移催化劑的用量、堿的選擇、水的用量控制以及反應時間和溫度等),取得了較為理想的效果,并結合制備型手性柱的超臨界流體色譜(SFC) 分離(SFC 是一種提高分離效率的綠色環保關鍵技術),以55%的分離產率、>99% ee 的光學純度制備了化合物56。
百時美施貴寶工藝小組仔細研究了在手性相轉移催化劑催化下進行不對稱環丙基化反應的機理[26],發現第一步烷基化反應并不是立體選擇的,而隨后的關環反應形成環丙烷則是立體選擇的( 圖14)。該小組分析了催化劑的降解途徑,通過對化學過程的透徹研究,采用切實可行的拆分- 重結晶和酯的直接酰胺化- 重結晶過程,開發出一條簡潔有效、不需要柱色譜分離的工藝( 圖15),有效制備了關鍵手性環丙烷α- 氨基酸中間體。值得強調的是,最開始反應得到的中間體為液態,但工藝研發人員一般偏愛處理固體物質( 因為其分離純化手段多,且易于放大操作),因此利用成鹽進行拆分、打漿提高純度以及隨后的酰胺化得到固體,不失為工藝綠化的良策。
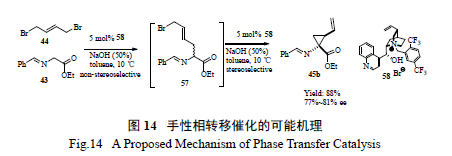
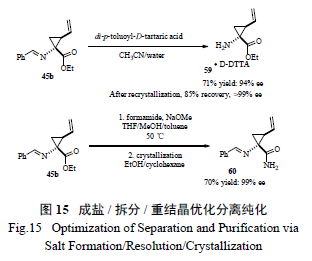
綜合上述3 家制藥公司對該反應系統的深入研究,不難發現其對關鍵反應的重視,同時也體會到反應及工藝綠色化是一個逐步完善的過程。綠色化方法如酶催化(BI)、SFC 分離技術(Merck),疊縮技術(BI) 和手性相轉移催化(Merck, BMS) 以及成鹽打漿(BMS, Merck, BI) 等實用技術都能得到很好地利用[27—29]。
4 烯烴復分解反應在合成大環內酯/ 內酰胺中的應用和工藝綠色化
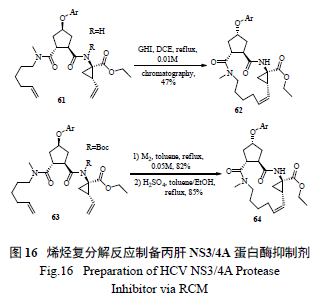
很多大環內酯和大環內酰胺藥物( 如克拉霉素和阿奇霉素) 都是重要的抗感染藥物,與普通百姓的健康息息相關。過去這類藥物的合成有的采用傳統的酯化和酰胺化,不僅產率、純度低,而且分離純化非常困難。近年來,烯烴復分解反應得到了廣泛利用,并逐漸成為常用的綠色化工藝手段和方法( 圖16)[30—31]。
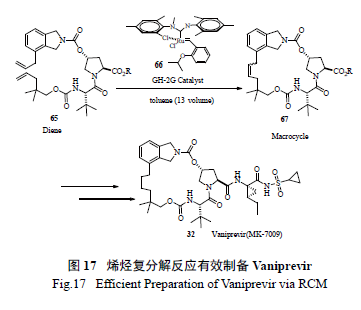
使用烯烴復分解反應最為成功的案例當屬Merck 制藥的工藝研究小組將其用于HCV 新藥研發,該小組釆用同時緩慢滴加Ru 催化劑和二烯底物的方法,極為出色地生產了藥物Vaniprevir(MK7009) [32]。該策略的亮點為:一是催化劑的用量很少(0.2 mol% ),二是底物濃度高(0.13 mol/L),不需要高度稀釋;三是極為優秀的高產率(91% )。這也是催化手段綠色工藝化學的極佳案例( 圖17)。
5 丙肝NS5A 蛋白酶抑制劑Elbasvir 關鍵反應的綠色化
ZEPATIER(Elbasvir / Grazoprevir) 是一款口服丙肝藥物組合[ 其中Elbasvir(71) 是一種NS5A抑制劑[33],Grazoprevir 是一種NS3/4A 蛋白酶抑制劑[34] ] 可有效抑制丙肝病毒的復制,2016 年1 月28 日被FDA 批準用于治療慢性丙型肝炎(HCV)基因1 型和4 型感染。該聯合藥物不需要聯合使用干擾素,避免了干擾素治療可能產生的嚴重不良反應。預計2017 年ZEPATIER 的銷售額有望達到15 億美元。圖18 是FDA 批準上市的NS5A 蛋白酶抑制劑的結構。
71 的發現路線如圖所示( 圖19),從簡單的鹵代苯酚出發,經過10 步反應,制得71 及其類似物,該法能夠方便快捷地合成一系列化合物,有效進行SAR 構效關系研究[35]。仔細考查71 結構,發現其中心結構(core) 兩邊所連接的基團是一樣的,如果中間體選擇合適,分子的局部對稱性對簡化合成是有幫助的。值得強調的是整個合成的重點是如何有效地制備光學化活性半縮醛胺(hemiaminal),這個結構在藥物分子中是極為罕見的。
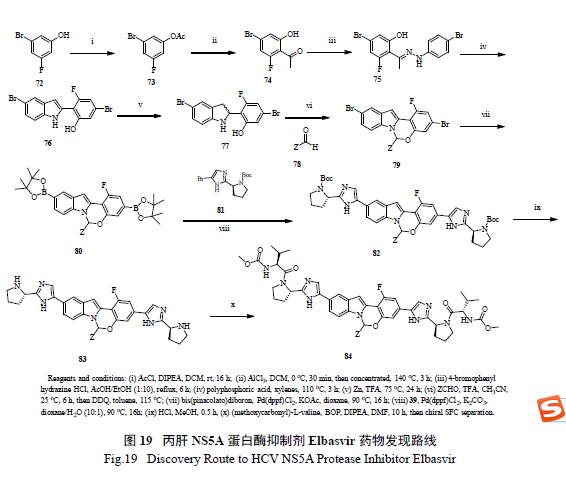
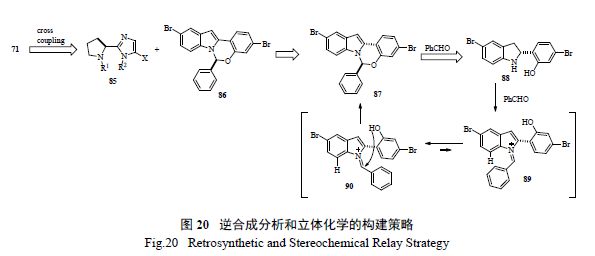
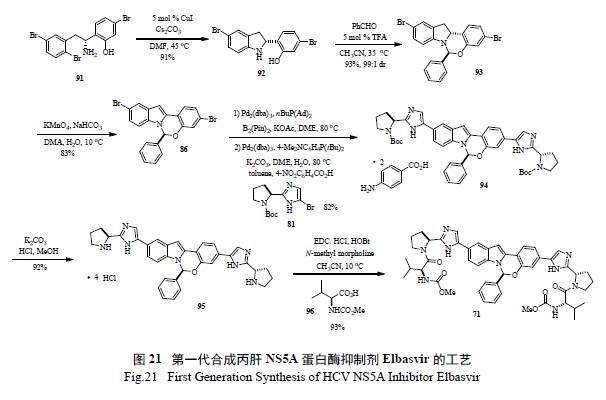
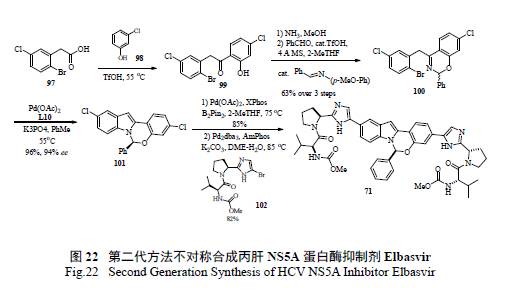
Merck 工藝研究人員通過逆合成分析( 圖20)和對反應機理的深度理解,認為最重要和最富有挑戰的是如何有效的構造半縮醛胺的手性中心。如圖21 所示,光學活性的二氫吲哚的制備,以及隨后的和醛加成消除制備活潑亞胺正離子都是至關重要的。這樣從光學活性的胺出發,經過9 步反應,有效制備了71[36]。美中不足的是,手性胺的制備是通過不對稱氫轉移來實現的,而通過氧化反應將二氫吲哚轉變為吲哚顯然有違于綠色化學的原理。在合成工藝研究中,對關鍵反應的攻關和突破對整個工藝的綠色化舉足輕重。Merck 工藝化學家們創造性地發展了一個新穎獨特的化學反應,高效地制備了光學活性的半縮醛胺[37]。如圖22 所示,其優秀之處表現在:(1) 釆用價廉易得的起始原料;( 2) 方便有效地制備外消旋苯并噁嗪,并通過其開環互變異構和手性雙膦單氧化物存在下的鈀催化Buchwald-Hartwig C-N 鍵的偶聯反應來控制半縮醛胺手性中心的形成;( 3) 通過對反應條件的廣泛篩選和優化,該關鍵反應的結果非常理想,ee 值高達94%,產率96%;(4) 避免了不必要的二氫吲哚和吲哚之間的轉換;( 5) 起始原料均為氯代苯而不是溴代苯,從原子經濟的原則來判斷后續的偶聯反應,也是更趨環保綠色的途徑。簡而言之,這是一條綠色化的工藝合成路線,必將成為合成與工藝綠色化的杰出案例。
6 結語與展望
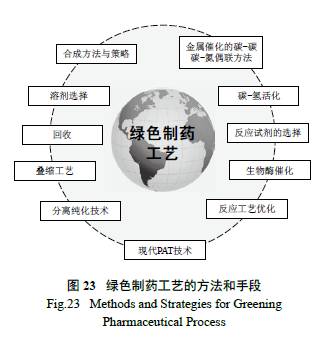
制藥工藝的綠色化是一個復雜、逐漸優化的系統工程( 圖23),是將一系列反應有效地“串聯”起來,從簡單易得的原料來制備復雜藥物分子的過程。涉及合成化學單元反應的有效連接,是一個“承上啟下”的科學活動[ 38]。因此,它不僅要考慮合成方法與策略、綠色的手段( 金屬催化的偶聯反應、C-H 活化、生物酶催化),而且追求實現完美工藝的技巧( 如工藝參數的優化、疊縮工藝的使用等)、分離的便利有效( 成鹽方式或打漿拆分) 以及溶劑的選擇與回收等。應該說,追求制藥工藝的完美和綠色化是業界同行的共識[39—40]。
最后,我們期待本文中分析總結的一些重磅新藥的綠色工藝的技巧和方法,能為業界同行的實際工作提供有益的借鑒與思考。同時希望國內制藥同仁不斷向跨國制藥的一流水平看齊,提高技術能力,開發出更加環保便捷、經濟實用的藥物綠色合成新方法、新工藝,為造福人類、創造更加文明的生態環境做出應有的貢獻。
致謝:本文是根據張霽博士在深圳南方科技大學舉辦的2016 中國制藥化學反應及工藝高峰論壇的報告整理而成。感謝張緒穆教授和周偉澄研究員給予的支持和建議
基金項目:廣東省引進創新創業團隊計劃(No.201301Y0105381261)
作者簡介:張 霽(1962—),男,博士,藥物研發/工藝研發首席科學家,曾任職于跨國藥業雅培(Abbott)、輝瑞(Pfizer)和百時美施貴寶(Bristol-Myers-Squibb),從事新藥研發和工藝研究。
聲明:化學加刊發或者轉載此文只是出于傳遞、分享更多信息之目的,并不意味認同其觀點或證實其描述。若有來源標注錯誤或侵犯了您的合法權益,請作者持權屬證明與本網聯系,我們將及時更正、刪除,謝謝。 電話:18676881059,郵箱:gongjian@huaxuejia.cn
