

 在線客服
在線客服
 快速發布
快速發布
 我的店鋪
我的店鋪
 我的化學加
我的化學加
 我的消息
我的消息
 我要充值
我要充值


 回到頂部
回到頂部



買產品

克難題

技術供需

求職招聘

發定制

項目整合

園區推薦

行業資訊

化學加智庫

商家
買產品
克難題
技術供需
求職招聘
發定制
項目整合
園區推薦
行業資訊
化學加智庫
商家
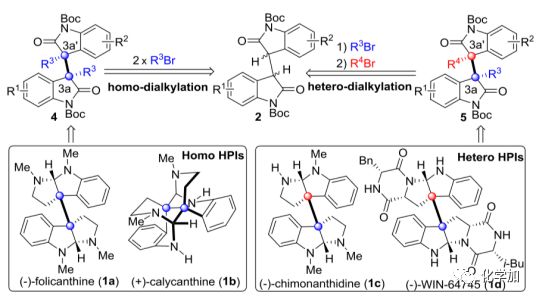
圖1代表性的HPI天然產物以及本文的合成設計
(圖片來源J. Am. Chem. Soc.)
如圖1所示,許多天然二聚和低聚的六氫吡咯吲哚(HPI)生物堿(包括同型和異型)都具有重要的生物活性,如抗真菌和抑制細胞生長作用等。從結構上講,這些分子通常在C3a和C3a’處包含一對具有空間位阻的季碳中心,從有機合成的角度來看,這是一個“艱巨的挑戰”。事實上,對于這種結構基元的催化不對稱合成非常困難,到目前為止僅有雙Michael加成和雙脫羧烯丙基化等方法,而且,這些報道的方法在收率和選擇性方面并不高,它們只適用于同型生物堿的合成。因此,非常有必要開發一種更高效和通用的催化不對稱二烷基化方法來合成這類結構基元。
在過去的幾十年中,四級銨、三唑鹽和相關的有機陽離子催化劑在2-氧化吲哚的單烷基化反應中起著重要作用。然而,對于二聚氧化吲哚同種和不同種二烷基化反應方面卻報道甚少,一是可能在第二個烷基化過程中C3a和C3a’之間的空間位阻作用增加,可能導致意想不到的副反應,如非對映異構的烷基化和C3a-C3a’鍵斷裂。另一個挑戰在于如何獲得不同類型的二烷基化反應(圖1),這需要兩種不同的親電試劑(R3Br和R4Br)的反應性和立體可控性要能足夠的匹配。因此,為了應對這些挑戰,基于前期的工作基礎,涂永強院士團隊發展了一種新型的SPA(螺環吡咯酰胺)-三唑陽離子催化劑,成功地實現了二聚氧化吲哚的不對稱同種和不同種二烷基化反應。
作者首先設計和制備了幾種新型的SPA-三唑溴化銨催化劑(Cat 1-6,圖2),并通過Cat 4的單晶證實了它們的絕對構象。以二聚氧化吲哚2a和4當量商業可得的溴乙酸芐酯3a為模型底物,研究了有機催化的不對稱二烷基化反應。通過對各種溶劑、堿和添加劑的篩選,最終發現在3 mol% Cat 1(entry 1,74%收率,93% ee,12.6:1 dr)的條件下,甲苯中室溫反應,就能較好地得到預期的同種二烷基化產物4aa。雖然其它催化劑(Cat 2-6)也能催化這一反應(entry 2-6),但結果較差。值得注意的是,N-甲基取代的Cat 6對映選擇性最差(-6% ee,entry 6),表明在該不對稱反應中,SPA型催化劑中N-質子的存在是立體選擇性的必要條件。
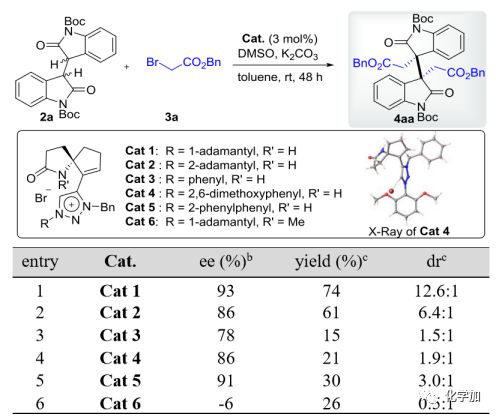
圖 2 同種二烷基化反應條件的篩選
(圖片來源 J. Am. Chem. Soc.)
得到最佳條件以后,作者對親電試劑R3Br和2a的同種二烷基化反應進行了底物擴展。圖3列出的結果表明,三組親電試劑Ⅰ-Ⅲ(Ⅰ組:溴乙酸芐酯3a-i,II組:溴乙酸烷基酯3j-l和Ⅲ組:烯丙基溴代物3m和3n,均為4倍當量)都成功地得到了預期的同種二烷基化產物4aa-4an,在大多數情況下(entry 1-14)都取得了滿意的結果。在第一組中,所有的親電試劑3a-i(entry 1-9)都能順利地反應,得到高對映選擇性(88-95% ee)和良好的產率(63-89%)。這些親電試劑的芐基取代基上電子效應對反應結果也有一定的影響,其中4-F-芐基酯3g的結果最好(entry 7)。對于第二組(entry 10-12),烷烴的位阻對反應效率的影響是非常明顯的。例如,小位阻的甲酯3j僅在2天內就能反應得到4aj,且具有良好的立體選擇性(entry 10),并由單晶確定了其結構和立體化學,而體積較大的乙基酯3k則需要更長的反應時間10天(entry 11)。此外,當將體積較大的正丁酯3l應用于該體系時,無法得到所需的產物4al(entry 12)。第三組中的烯丙基親電試劑3m和3n對此反應也是可行的,但結果卻不相同,3m的反應具有較高的ee和產率(entry 13),而3n則具有較高的dr和產率(entry 14)。
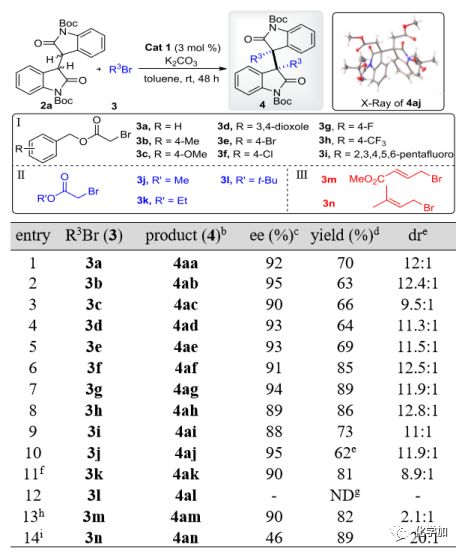
圖 3 同種二烷基化中親電試劑的考察
接著,作者以3a為親電試劑,對多種芳環上單、雙取代的二聚氧化吲哚類化合物2進行了考察。如圖 4所示,該反應具有廣泛的底物兼容性。對于C5a、C6a或C7a的單取代底物,吸電子取代基(2-5,7-9和12)比給電子取代基普遍有利,然而,C4a-Cl取代的底物不能得到所需產物,只能產生復雜的混合物,這可能是由于其對反應中心(C3a或C3a’)的空間屏蔽所致。在雙取代底物(entry 13-18)中,非對稱(entry 13-15)和對稱(entry 16-18)雙取代在此反應中都是有效的,且具有優良的對映選擇性(92-98% ee)、良好到優良的非對映選擇性(7.1:1至>20:1 dr)以及中等至良好的產率(46-85%)。與單取代底物類似,二取代物的電子性質對反應結果也有顯著影響。例如,二氯代底物能快速反應生成相應的產物4ra,產率為85%,ee 值為92% (entry 17),而二甲氧基取代底物反應的結果相對較差(33%產率,98% ee,>20:1 dr),還需要添加DMSO作為極性共溶劑進一步改進,才能以較高的產率得到4sa(78%產率,92% ee,18:1 dr,entry 18)。重要的是,對于產物4ca和4da,可以通過C-C偶聯反應和進一步轉化來合成更復雜的三聚HPI生物堿,具有重要的應用價值。
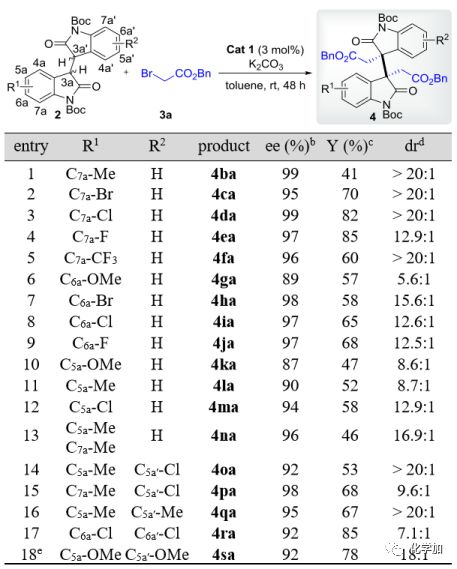
圖 4 同種二烷基化反應中二聚氧化吲哚底物的擴展
完成了二聚氧化吲哚的同種二烷基化反應,接著作者用兩種不同的親電試劑R3Br和R4Br,研究了更具挑戰性的一鍋法不同種的二烷基化。最初作者發現在Cat 1(3 mol%)催化下, 1當量的3a和2a反應能得到單烷基化產物,化學選擇性良好,產率約85%,沒有檢測到二烷基化產物。根據這一觀察結果,在2a消失后,向反應體系中加入3當量的3g,就得到了所需的不同種二烷基化產物5a,產率54%,ee值96%。受此鼓舞,使用不同的親電試劑R3Br, 作者又嘗試了更多的不同種二烷基化反應,均能得到預期的產物5b-f。特別地,5c-f可作為關鍵中間體,用于合成復雜的HPI生物堿。
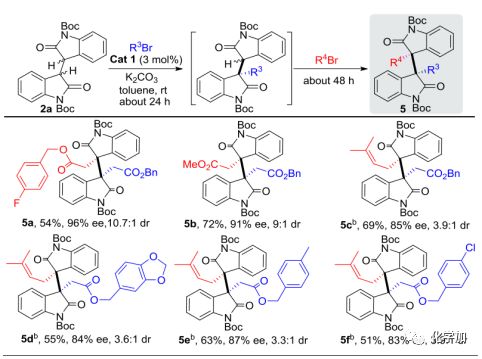
圖 5 2a的不同種二烷基化反應研究
接著,作者對三唑催化的烷基化反應過程中的立體化學控制進行了探究,根據觀察到的結果,作者提出了單烷基化和二烷基化反應的親核進攻模型(圖 6)。由于Cat 6沒有酰胺N-H部分,僅給出了較差的立體選擇性。而在Cat 1和烯醇式反應時,每一步烷基化過程中都可能存在氫鍵和離子對相互作用,由于金剛烷基大的位阻作用,中間體從背面向親電試劑的親核進攻是不利的,從而導致主要形成(S,S)-二烷基化產物。
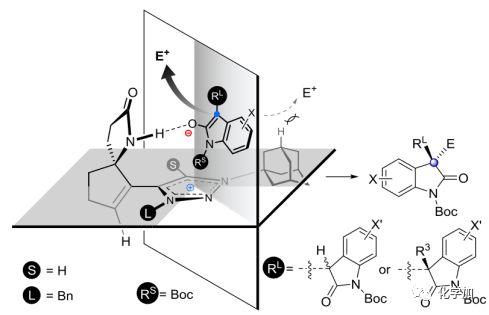
圖 6 立體化學控制模型
為了驗證該催化不對稱二烷基化反應的有效性,作者還將其用于了天然產物的全合成中,實現了(–)-folicanthine (1a)的形式全合成和(–)-chimonanthidine (1c)的首次不對稱全合成。在(1c)的合成中,主要挑戰在于在兩個吡咯烷環存在下,如何選擇性地用甲基僅保護一個N-H。而用不同種二烷基化產物5c(可以克級規模制備,重結晶后ee大于99%)作為關鍵中間體,只需要一次柱層析純化,經脫Boc、甲基保護、胺解及還原胺化就可以有效地得到關鍵的合成子7,具有81%的總收率,區域選擇性還原實現了吲哚部分的胺化/環化反應,隨后化合物7的C=C鍵氧化裂解,得到醛化合物8,通過還原胺化反應得到產物9,再一次還原胺化/環化反應生成第二個吡咯烷環,最后在Pd(OH)2/C催化條件下脫去芐基保護基,就實現了(–)-chimonanthidine (1c)的首次不對稱全合成。
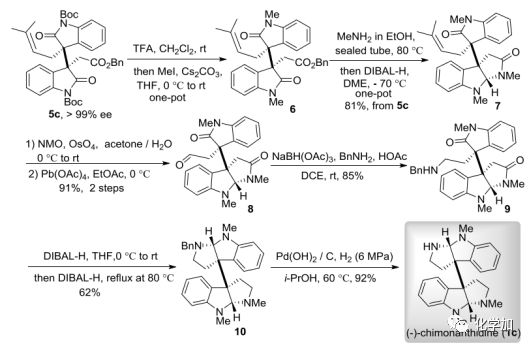
圖 7(–)-chimonanthidine的不對稱全合成
總結:涂永強院士團隊成功地基于氮雜螺環骨架開發了SPA三唑陽離子催化劑,并利用該催化劑實現了二聚氧化吲哚類化合物的不對稱同種和不同種二烷基化反應,該反應操作簡便、條件溫和,可以高效地構建極具挑戰性的相鄰季碳中心(收率高達82%,ee高達99%,具有>20:1 的dr)。到目前為止,該轉化是構建這類合成基元的最有效的方法。此外,從產物5c出發,可以很容易地實現(–)-chimonanthidine (1c)的不對稱全合成,體現了該方法的重要應用價值。
聲明:化學加刊發或者轉載此文只是出于傳遞、分享更多信息之目的,并不意味認同其觀點或證實其描述。若有來源標注錯誤或侵犯了您的合法權益,請作者持權屬證明與本網聯系,我們將及時更正、刪除,謝謝。 電話:18676881059,郵箱:gongjian@huaxuejia.cn
