

 在線客服
在線客服
 快速發布
快速發布
 我的店鋪
我的店鋪
 我的化學加
我的化學加
 我的消息
我的消息
 我要充值
我要充值


 回到頂部
回到頂部



買產品

克難題

技術供需

求職招聘

發定制

項目整合

園區推薦

行業資訊

化學加智庫

商家
買產品
克難題
技術供需
求職招聘
發定制
項目整合
園區推薦
行業資訊
化學加智庫
商家

(圖片來源:J. Am. Chem. Soc.)
對映體富集的α-三級胺是藥物和天然產物中的一個重要的結構單元(Figure 1A)。雖然它們可以使用手性助劑進行制備,但催化不對稱方法尚不成熟。同時,三級硝基烷烴也可通過硝基的還原,合成相應的三級胺。因此,立體選擇性制備三級硝基烷烴的方法非常有吸引力,但仍不發達,通常涉及過渡金屬催化的烯丙基烷基化或有機催化的Michael加成和Aza-Henry反應。此外,二級硝基化合物(nitronates)與烷基鹵化物的C-烷基化是構建全取代硝基烷烴的一個潛在的策略,但由于競爭性的O-烷基化易形成羰基副產物。目前,化學家們發現,具有特定缺電子親電試劑的單電子轉移(SET)機理,有利于進行C-烷基化反應。Watson課題組通過自由基機理實現了一系列通用的過渡金屬催化(Cu或Ni)硝基化合物與簡單烷基鹵化物親電試劑的C-烷基化反應。隨后,Watson課題組報道了一種鎳催化一級硝基化合物的不對稱C-烷基化反應,合成了對映體富集的二級硝基烷烴。然而,對于通過硝基化合物與烷基鹵化物進行的不對稱C-烷基化構建三級硝基烷烴的方法,目前尚未有相關的報道。作者設想,酶是否能夠催化硝基化合物進行不對稱C-烷基化反應以生成三級硝基烷烴。與酶催化相關的高度選擇性和進化性,使其成為一種具有吸引力的策略。盡管在各種生物催化的共軛加成或Henry反應中使用了硝基化合物作為親核試劑,但沒有一種方法能夠構建手性三級硝基烷烴。值得注意的是,天然酶不能夠催化硝基烷烴與烷基鹵化物的C-烷基化反應。因此,作者設想,需要一種非天然的催化機理來應對這一挑戰。2022年,Hyster課題組(Nature 2022, 610, 302.)報道了黃素依賴性“烯”還原酶(EREDs)可以催化烷基鹵化物和硝基烷烴之間的不對稱交叉親電偶聯(XEC)。該反應涉及烷基自由基的形成,可與硝基化合物反應形成硝基陰離子。酶介導的裂解(mesolytic cleavage)形成一個三級自由基,可以通過氫原子轉移(HAT)立體選擇性地淬滅。近日,美國康奈爾大學Todd K. Hyster課題組報道了一種有助于硝基自由基陰離子氧化以合成三級硝基烷烴的酶的鑒定和進化(Figure 1B)。烷基鹵化物1的光誘導還原可生成烷基自由基4,可與原位生成的硝基化合物5反應,實現C-C鍵的立體選擇性構建,生成硝基陰離子6,6可通過單電子氧化終止,獲得所需的C-烷基化的硝基烷烴產物3。首先,作者以α-氯代酰胺1a與(2-硝基丙基)苯2a作為模型底物,進行了相關反應條件的篩選(Figure 1C)。當GkOYE作為催化劑,Tricine作為緩沖液(100 mM,pH 9.0),DMSO作為助溶劑,可在青色LED照射下室溫反應24 h,可以56%的收率得到硝基烷烴產物3a,er為78:22。

(圖片來源:J. Am. Chem. Soc.)
為了進一步提高反應的效率,作者進行了蛋白質工程(Figure 2)。研究表明,當使用三重突變體(D73C/A104H/Y264W,即GkOYE-G7)時,可獲得96%收率與96:4 er的產物3a。

(圖片來源:J. Am. Chem. Soc.)
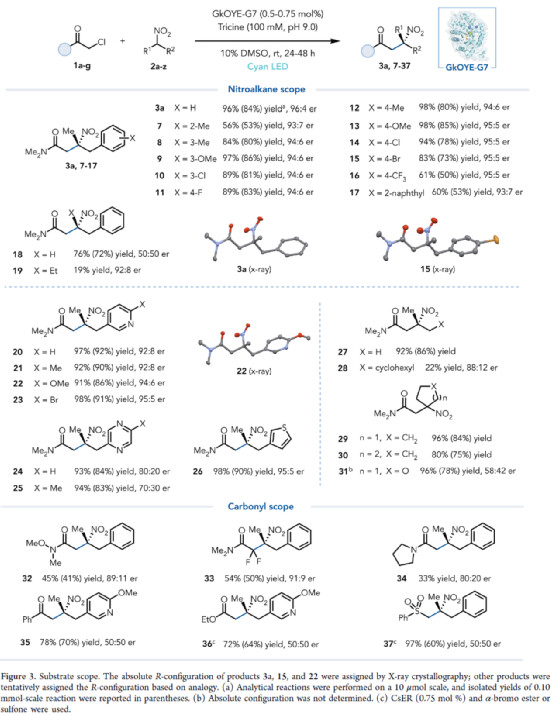
在獲得上述最佳反應條件后,作者對底物范圍進行了擴展(Figure 3)。研究表明,一系列不同取代的硝基烷烴,均可順利進行反應,獲得相應的產物3a與7-31,收率為19-98%,er為50:50-96:4。同時,三級酰胺、α-鹵代酮、α-鹵代酯或砜,也是合適的底物,獲得應的產物32-37,收率為33-97%,er為50:50-91:9。
(圖片來源:J. Am. Chem. Soc.)
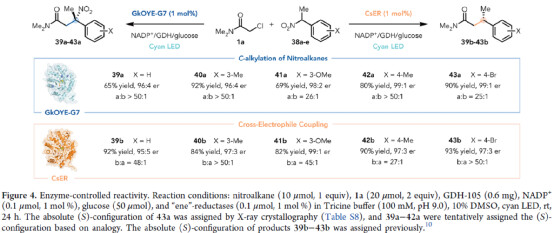
緊接著,作者對酶控反應性進行了研究(Figure 4)。研究表明,當使用GkOYE-G7催化體系時,反應可進行相應的C-烷基化反應,獲得相應的產物39a-43a,收率為65-92%,er為96:4-99:1,a:b為25:1->50:1。當使用CsER催化體系時,反應還可進行相應的不對稱交叉親電偶聯,獲得相應的產物39b-43b,收率為82-93%,er為95:5-99:1,b:a為27:1->50:1。
(圖片來源:J. Am. Chem. Soc.)
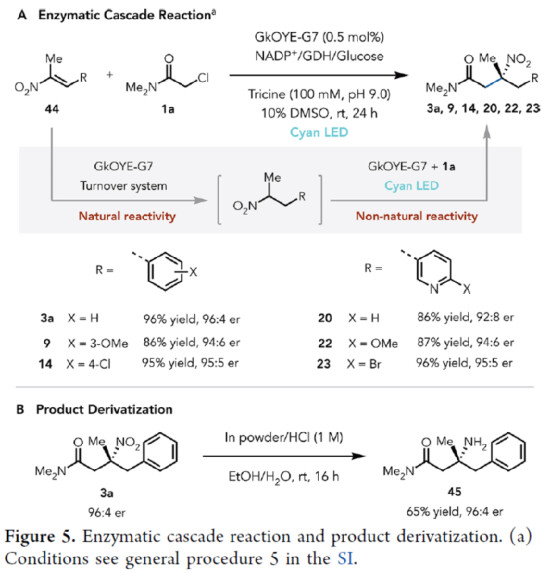
此外,作者以GkOYE-G7作為生物催化劑,通過將氫化物轉移機理(天然催化活性)與光氧化還原自由基機理(非天然催化活性)相結合,可實現一系列硝基烯烴的烷基化反應,獲得相應的產物3a、9、14、20、22和23,收率為86-96%,er為92:8-96:4(Figure 5A)。其次,3a可通過進一步的還原,可以65%的收率獲得胺化產物45,er為96:4(Figure 5B)。
(圖片來源:J. Am. Chem. Soc.)
最后,作者對反應機理進行了研究(Figure 6)。首先,GkOYE-G7在tricine緩沖液中于青色光照射下,通過UV-vis光譜監測發現,反應形成了FMNhq(flavin hydroquinone)和FMNsq(flavin semiquinone)的混合物(Figure 6A)。1a與2a在無光的條件下,未能發生反應,表明了基態FMNsq和基態FMNhq均不負責自由基的引發。同時,基態FMNhq、基態或激發態FMNhq均不能作為還原劑。通過進一步的研究表明,電荷轉移(CT)配合物物負責α-氯代酰胺1a的還原(Figure 6B)。

(圖片來源:J. Am. Chem. Soc.)
總結
美國康奈爾大學Todd K. Hyster課題組報道了一種光酶催化硝基烷烴的不對稱C-烷基化反應 ,合成了一系列手性三級硝基烷烴。其中,該反應通過工程化的ERED(GkOYE-G7)實現,其特征是通過對映匯聚性Csp3-Csp3鍵的形成實現了四取代中心的構建。雖然進化的GkOYE-G7對催化硝基烷烴的C-烷基化表現出高度的專一性,但其仍保留了天然的還原反應性,通過一種獨特的單酶雙機理串聯反應,從而實現了從易得的硝基烯烴合成手性三級硝基烷烴,該策略還解決了過渡金屬催化硝基烷烴不對稱C-烷基化中的長期挑戰,擴大了生物催化的邊界。
文獻詳情:
Haigen Fu, Tianzhang Qiao, Jose M. Carceller, Samantha N. MacMillan, Todd K. Hyster*. Asymmetric C?Alkylation of Nitroalkanes via Enzymatic Photoredox Catalysis. J. Am. Chem. Soc. 2023, https://doi.org/10.1021/jacs.2c12197
聲明:化學加刊發或者轉載此文只是出于傳遞、分享更多信息之目的,并不意味認同其觀點或證實其描述。若有來源標注錯誤或侵犯了您的合法權益,請作者持權屬證明與本網聯系,我們將及時更正、刪除,謝謝。 電話:18676881059,郵箱:gongjian@huaxuejia.cn
