

 在線客服
在線客服
 快速發布
快速發布
 我的店鋪
我的店鋪
 我的化學加
我的化學加
 我的消息
我的消息
 我要充值
我要充值


 回到頂部
回到頂部



買產品

克難題

技術供需

求職招聘

發定制

項目整合

園區推薦

行業資訊

化學加智庫

商家
買產品
克難題
技術供需
求職招聘
發定制
項目整合
園區推薦
行業資訊
化學加智庫
商家
柴油是遠程運輸和工程機械領域不可或缺的高能量密度燃料,商品柴油需要預脫硫以減少硫氧化物排放。目前,石化行業采用加氫脫硫實現上述目的,需要高昂的成本和能量消耗。利用空氣作為可持續的氧化劑,分子氧氧化脫硫(AODS)是加氫脫硫的理想替代。然而,AODS需要在低溫和常壓條件下實現硫化物的轉化,以降低能耗和爆炸風險。因此,需要高效催化劑克服溫和條件下O2活化的挑戰。
迄今,以非金屬W、Mo和V、金屬Cu和Pt等為主體的多相催化劑對噻吩類硫化物的氧化反應活性較高。其中,Mo基金屬氧化物兼具良好的催化活性和優異的催化穩定性。三氧化鉬本身是非活性的,需要通過與其他氧化物復合來引入穩定的低價態Mo,誘導表面氧空位形成,增強對O2的吸附活化。盡管學者們致力于調控Mo位點的結構和化學環境,但難以對Mo位點電子結構進行精準調控和優化。
針對該問題,北京大學郭少軍團隊提出了一種在金屬氫氧化物納米片(Mg,Fe,Co,Ni,Cu和Zn)上錨定Mo單原子的通用性方法,合成過程簡單且易于規模化。通過改變宿主氫氧化物,可以精準地調控Mo單原子位點的電子結構。基于此,設計出一種亞納米厚度的氫氧化鈷納米片負載原子級鉬催化劑(Mo/Co(OH)2),并將其用于噻吩硫化物的高效分子氧氧化反應。該催化劑的轉換頻率比目前最先進的多金屬氧化物高出兩個數量級,可在60 °C的低溫條件下激活反應,并在80 °C下實現加氫柴油的完全脫硫,能耗低(如果使用煉油廠余熱,則不會消耗額外能源)、耐用性高,進一步溶劑萃取可使脫硫柴油的硫含量達到當前的硫含量標準(<10 ppm)。
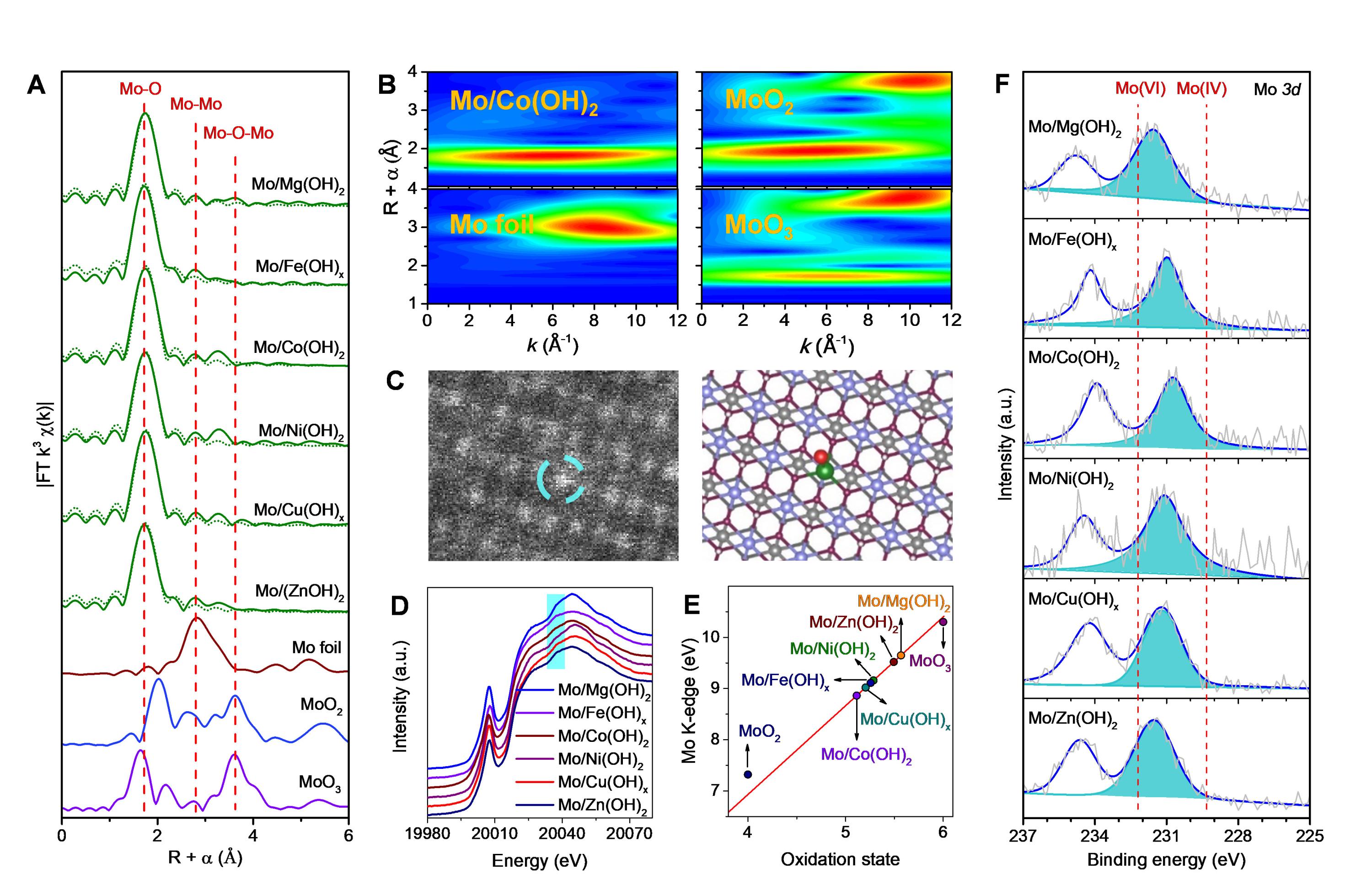
圖1. Mo單原子位催化材料的電子結構與配位環境表征。
作者發現,Mo原子位的電子狀態可通過宿主氫氧化物進行精確調節,導致宿主的電子給體效應與催化性能之間存在強烈的相關性。作者建立一個通用描述符——宿主材料的功函數,可很好地解釋宿主材料對催化性能的影響。具體而言,主體的功函數影響從催化劑到O2的電荷轉移的熱力學有利性,進而決定O2是否可以在低溫下被激活。如果物理吸附中性O2分子的O2親和勢高于催化劑的功函數,則會發生自發的電荷傳輸。否則,該反應需要額外的能量來完成電子轉移,這意味著更高的反應溫度。作者通過紫外光電子能譜(UPS)證實,主體材料的功函數與Mo單原子的催化性能存在相關性。
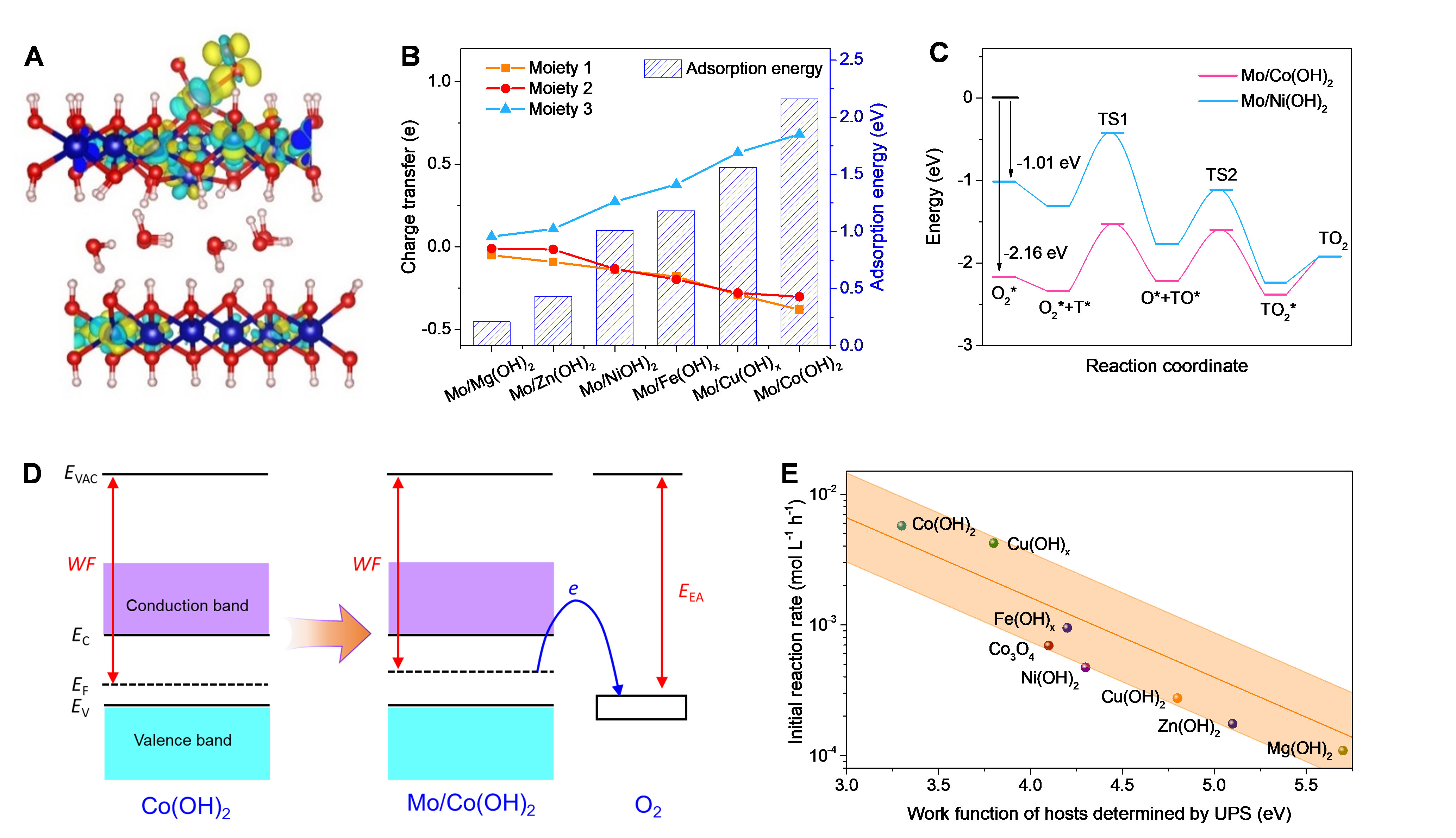
圖2. Mo基單原子位催化材料的噻吩類硫化物分子氧氧化催化機理。
作者對Mo基單原子位催化材料開展了應用性能評價,在80 °C下對加氫柴油和直餾柴油進行了AODS實驗。對于加氫柴油而言,幾乎所有的硫化物均在50 min內完全轉化(圖3A),這表明Mo基單原子位催化材料具有優異的催化活性。圖3B揭示了該Mo基單原子位催化材料在經過多次催化循環測試后仍可保持良好的轉化效率。通過進一步對重復使用的催化材料進行XPS、Raman、XANES和EXAFS表征,可以證實Mo位點的精細結構或氧化狀態均未發生變化(圖3C-D),表明Mo基單原子位催化材料具有優異的催化穩定性。
總之,Mo/Co(OH)2可在80°C下實現加氫柴油的完全脫硫,能耗低(如果使用煉油廠余熱,則不會消耗額外能源)、耐用性高,進一步溶劑萃取可方便地使脫硫柴油的硫含量達到當前的硫含量標準(<10 ppm)。
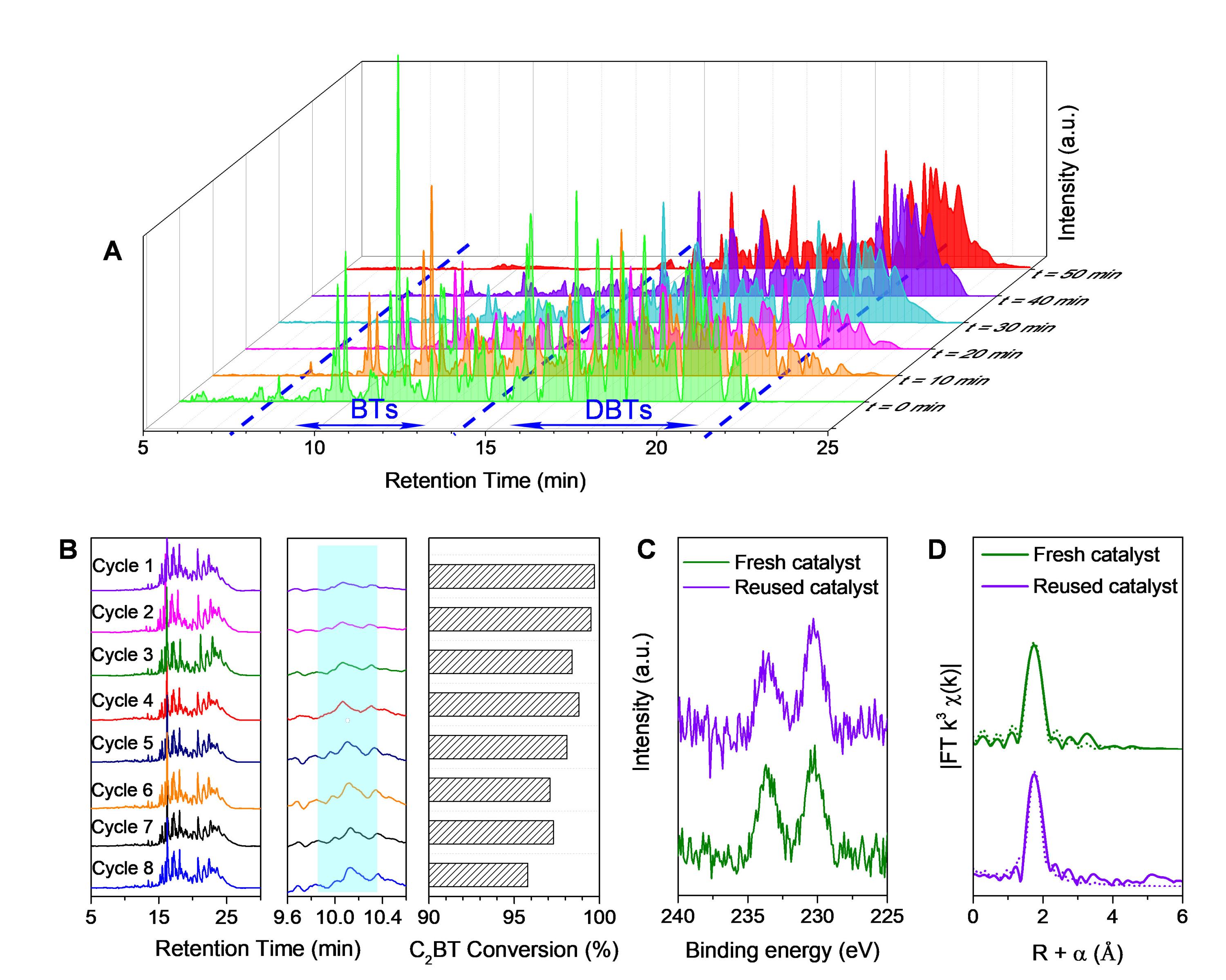
圖3. Mo單原子位催化劑在柴油AODS轉化中的實際應用性能。
聲明:化學加刊發或者轉載此文只是出于傳遞、分享更多信息之目的,并不意味認同其觀點或證實其描述。若有來源標注錯誤或侵犯了您的合法權益,請作者持權屬證明與本網聯系,我們將及時更正、刪除,謝謝。 電話:18676881059,郵箱:gongjian@huaxuejia.cn
