

 在線客服
在線客服
 快速發布
快速發布
 我的店鋪
我的店鋪
 我的化學加
我的化學加
 我的消息
我的消息
 我要充值
我要充值


 回到頂部
回到頂部













近日,西湖大學石航教授課題組在JACS上報道了一種銠催化的苯酚胺化,提供了各種苯胺的簡潔合成,其中水是唯一的副產物。親芳銠催化劑通過π-配位促進苯酚固有的困難的酮-烯醇互變異構化,允許隨后與胺的脫水縮合。作者通過對具有各種電性的大量酚類和多種伯胺和仲胺進行反應,證明了這種氧化還原中性催化的普適性。結構復雜的生物活性分子(包括藥物)的后期功能化進一步說明了該方法潛在的廣泛用途。文章DOI:10.1021/jacs.1c12622
苯胺是功能性材料、醫藥、農產品和天然產物中常見的結構單元,因此,開發用于制備具有苯胺結構單元的化合物的有效方法是合成化學中的一項重要工作。在過去的幾十年中,使用鈀、銅或鎳催化劑,在過渡金屬催化的芳基鹵化物和芳基硼酸等芳香族化合物的胺化領域取得了長足的進步。然而,酚的胺化仍然難以捉摸。由于Ph-OH鍵的高解離能(> 450 kJ/mol)、酚羥基對氧化等轉化的敏感性以及它們對過渡金屬質子化的傾向,酚類不適合過渡金屬-催化的交叉偶聯反應。在過渡金屬催化的C-N鍵形成反應(包括Buchwald-Hartwig胺化和Ni-催化的胺化)中,三氟甲磺酰基等活化單元對于促進C-O鍵的氧化加成是必不可少的(圖1a)。下載化學加APP,閱讀更有效率。
圖1. 從苯酚合成苯胺(圖片來源:J. Am. Chem. Soc.)
在制定苯胺的簡明合成策略時,作者借鑒了廣泛應用于有機催化、還原胺化和Mannich反應的羰基胺縮合反應;他們的想法是,如果縮合可以與苯酚固有的困難的酮-烯醇互變異構化相結合,就可以開發出直接獲得苯胺的方法。互變異構平衡取決于酮和烯醇互變異構體的相對穩定性(圖1b)。對于丙酮,在正常條件下,平衡強烈有利于酮的形成,而苯酚的二烯酮互變異構體的形成由于失去芳香性而被完全抑制。1904年,Bucherer報道了亞硫酸氫鈉促進的萘酚胺化,其中萘酚可以互變異構為烯酮,因為稠環去芳構化的能壘相對較低。盡管苯胺可以通過使用非均相催化劑的優雅氫化去芳構化/脫氫芳構化序列從酚類和胺類中生成,但外部或內部氫源是必不可少的。此外,苯酚與胺的胺化可以通過一系列氧化去芳構化、縮合和氧化還原異構化來完成。
鑒于Lewis酸可以推動酮-烯醇平衡向烯醇化物方向發展,作者想知道酸是否會特異性地激活酚環以迫使形成二烯酮而不是酚鹽,從而為與胺的脫水縮合提供機會。在以往對金屬-芳烴π-配位配合物的研究中,發現η6-苯酚配合物由于金屬的吸電子作用容易去質子化,生成酮形式的η5-苯氧基配合物。然而,這種反應在催化中的應用仍有待證明。
在此,作者報道了一種無需活化試劑或還原劑/氧化劑的銠催化酚與胺直接胺化的有效方法(圖1c)。該方法在原子經濟性、酚類可再生性、以及廢棄物等方面可以滿足“綠色”。作者設想這種氧化還原中性催化將通過以下機制進行。首先,親芳性的Rh(III)催化劑會通過π-配位與苯酚連接,去質子化后產生瞬時的η5-苯氧基配合物II;II的羰基胺縮合得到亞胺中間體III,它會發生互變異構,然后進行芳烴解離以產生所需的苯胺產物。
首先,作者以4-甲基苯酚(1a)和哌啶(2a)之間的模型反應開始了研究(圖2a)。通過廣泛的條件篩選,發現 [Cp*RhCl2]2(催化劑1)在含有10 mol% Na2CO3的庚烷中,在120 °C下得到所需的產物3a,收率為20%。將Cp*環上的一個甲基替換為氫(催化劑2)或脂肪族基團,如環己基(催化劑3)、異丙基(催化劑4)或三氟甲基(催化劑5),降低了產率。相反,具有芳族取代基的催化劑(6-10)的產率與使用催化劑1的產率相似或更好。特別是10,具有3,5-二(三氟甲基)-苯基,產率為40%。將反應溫度提高到140 ℃,收率提高到80%,加入分子篩使收率提高到91%。使用預先形成的銠單體[CpCF3Rh(MeCN)3]2+ (催化劑11),在不需要 Na2CO3的情況下得到70%的3a。
有了最佳條件,作者探索了苯酚與哌啶2a的反應范圍(圖2b)。廣泛的對位取代苯酚是合適的,以高達90%的產率提供所需的產物3b-3y。電中性(3b-3l)、給電子(3m-3p)和吸電子(3q-3t)基團和芳香環(3u-3y)均耐受。無論取代物(3z-3ae)的電性如何,一系列間位取代的苯酚都經歷了所需的反應。此外,多取代苯酚(3af-3ah)和稠環苯酚(3ai-3ao)的胺化反應以中等至良好的收率平穩地提供苯胺。值得注意的是,雜環部分,包括吡啶(3y)、二噁英(3ak)和四氫喹啉(3am和3an),在反應條件下都是可耐受的。胺化對羥基的空間位阻敏感,2-甲基苯酚僅產生痕量所需產物(3ap)。
圖2. 條件篩選及苯酚的底物范圍(圖片來源:J. Am. Chem. Soc.)
接下來,作者通過與4-甲基苯酚(1a)進行反應來評估胺的反應范圍(圖3)。各種帶有直鏈(3aq-3ax)、β-支鏈(3ay-3bc)或α-支鏈(3bd-3bi)取代基的伯胺都是合適的,以40-89%的產率提供相應的苯胺。此外,芐胺(3bk)和各種衍生物(3bl-3bp)在貴金屬存在下易于β-H消除,也與反應條件相容。本文的方法也適用于廣泛的仲胺。具體來說,藥物中常見的哌啶(3bq-3cd)和哌嗪(3ce-3cl)是優良的底物,以及一系列反應性官能團,包括腈(3bq)、羧酸酯(3bs)、1°-3 °醇(3bt,3bu,3by),氨基(3bv,3ce-3cg),酰胺(3cd),氨基甲酸酯(3bw,3ca),吡啶基(3cj,3ck)和嘧啶基(3cl)都是耐受的。兩個螺環氮雜環丁烷(3cm, 3cn)的成功胺化進一步證明了該方法的相容性。此外,5至7元環狀和橋環胺,包括吡咯烷(3co-3ct)、嗎啉(3cu)、四氫異喹啉(3cv)、氮雜環庚烷(3cw)和二氮雜雙環[2.2.1]庚烷(3cx)也是適合的,以高達91%的收率提供相應的產品。無環仲胺以中等收率產生所需的產物(3cy-3de)。然而,由于空間位阻,叔丁胺(3bj)和二異丙胺(3df)未能進行所需的N-芳基化。
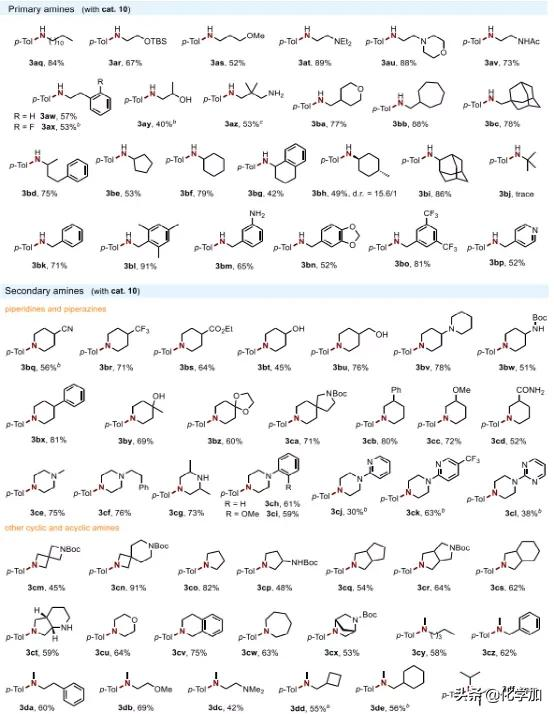
圖3. 胺的底物范圍(圖片來源:J. Am. Chem. Soc.)
該反應的高度相容性鼓勵作者研究其在后期功能化中的實用性(圖4a)。Paracetamol(撲熱息痛)是一種常見的鎮痛解熱藥,hordenine(大麥牙堿)可緩解支氣管炎癥狀,它們都是合適的底物,分別提供4a和4b。雙酚A的胺化提供了70%的單胺和二胺產物(4c)。縮酮-雌酮和17-庚酸雌二醇也與反應條件相容,分別得到4d和4e。此外,該方法成功地用于一系列藥物的 N-芳基化,包括伯胺美西律(4f)和脫氫樅胺(4g)、無環仲胺氟西汀(4h)、哌啶帕羅西汀(4i)和哌嗪阿莫沙平(4j)。
圖4. 后期官能團化及機理研究(圖片來源:J. Am. Chem. Soc.)
為了深入了解機理,作者監測了4-甲基苯酚(1a)和哌啶(2a)之間的催化胺化反應,并觀察到 2a的逐漸消失和苯胺3a的形成;沒有檢測到任何亞胺、環己酮或環己烯-1-酮(圖4b)。此外,1a與α-氘代十二胺(2aq)胺化生成苯胺3aq(95%氘摻入),在12小時內氘摻入沒有明顯衰減;然而,當反應進行24-36小時(圖4c)時,觀察到衰減。值得注意的是,沒有檢測到芳族H/D交換,表明這種胺化的氫轉移機制不太可能。當η5-phenoxo絡合物Rh-1由相應的Cp*Rh η6-苯酚絡合物在堿存在下通過易于去質子化形成的,經受胺化條件(圖4d),以55%的產率獲得游離苯胺3af和3,5-二甲基苯酚(1af)。此外,η6-苯胺絡合物Rh-2和1af之間的芳烴交換反應以幾乎定量的產率產生η5-苯氧絡合物Rh-1和游離苯胺。這些結果支持圖1c中描述的π配位激活機理。
總結:西湖大學石航教授課題組開發了一種在沒有外源活化試劑的情況下通過銠催化的酚和胺之間的脫水縮合反應簡明合成苯胺的新策略。鑒于該方法在苯酚和胺方面的廣泛范圍及其廣泛的官能團耐受性,將具有廣泛的合成效用。